Síndrome miastênica congênita (SMC) é um distúrbio neuromuscular hereditário causado por defeitos de diversos tipos na junção neuromuscular. Os efeitos da doença são semelhantes aos da síndrome de Lambert-Eaton e da miastenia gravis, sendo a diferença que a SMC não é um distúrbio autoimune. Existem apenas 600 casos familiares conhecidos deste distúrbio e estima-se que sua frequência geral na população humana seja de 1 em 200.000.
Introdução
O que você precisa saber de cara
Visão geral
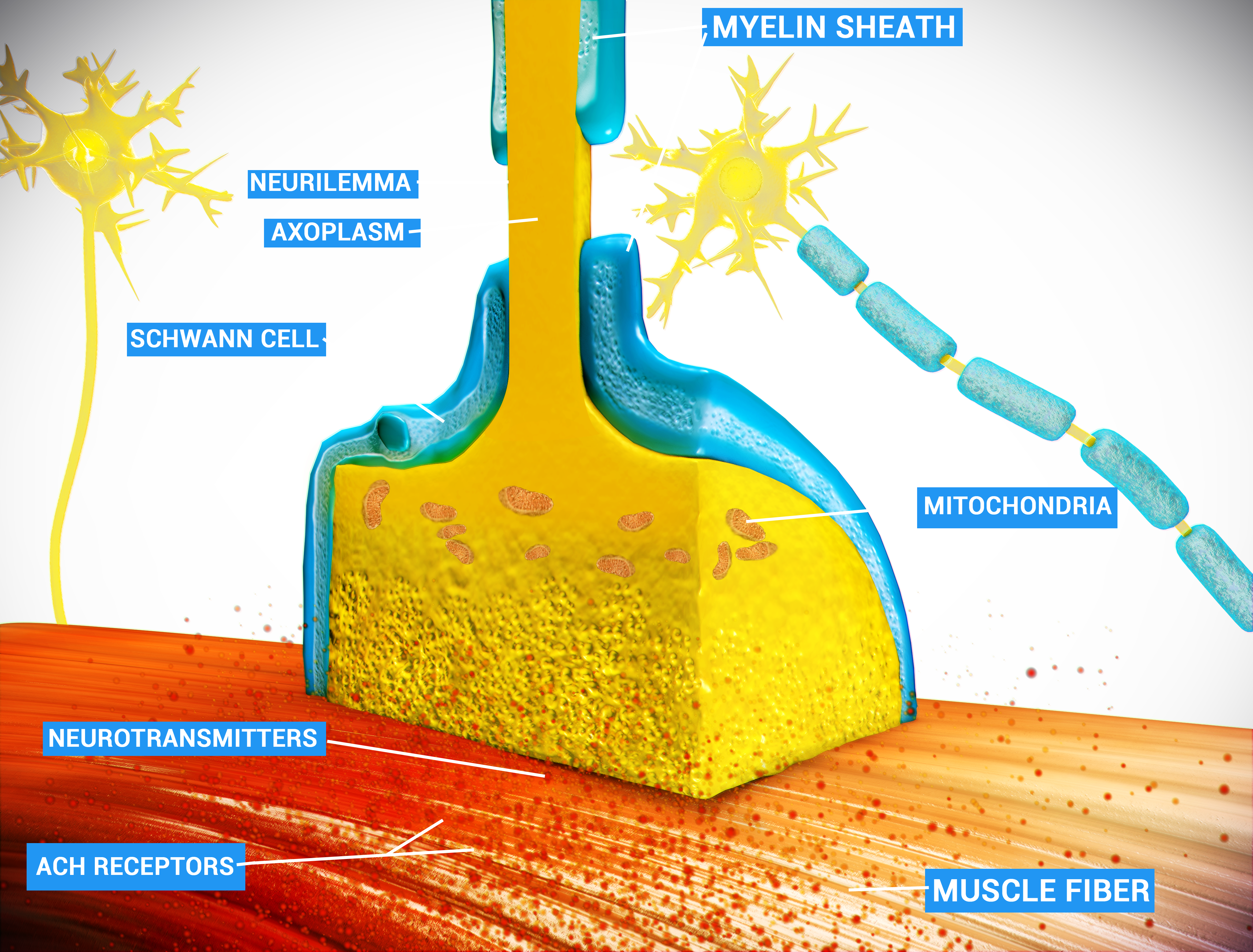
As síndromes miastênicas congênitas pré-sinápticas são um grupo de doenças neuromusculares hereditárias raras que afetam a transmissão do impulso nervoso do nervo para o músculo. O termo 'pré-sináptico' indica que o defeito genético está localizado na parte do neurônio que libera o neurotransmissor (acetilcolina) na junção neuromuscular. Essas condições geralmente se manifestam desde o nascimento (período neonatal) ou na primeira infância, causando fraqueza muscular que piora com o esforço (fadiga).[1][4]
Sinais e sintomas
Os sintomas podem variar de pessoa para pessoa, mas frequentemente incluem fraqueza muscular generalizada, fadiga fácil (fraqueza fatigável) e fraqueza que afeta tanto os músculos próximos ao tronco (proximal) quanto os das extremidades (distal). Muitos pacientes apresentam episódios intermitentes de insuficiência respiratória devido à fraqueza muscular, apneia central do sono, desconforto respiratório episódico e parada respiratória. Outros sinais comuns são arreflexia (ausência de reflexos), polineuropatia motora, olhar fixo, marcha na ponta dos pés, estridor (ruído ao respirar), episódios de engasgos, regurgitação nasal, deformidades da coluna vertebral, rigidez espinhal, atrofia da fibra muscular, amiotrofia distal (perda de massa muscular nas extremidades) e mandíbula estreita.[1][4]
Causas genéticas
Estas síndromes são causadas por mutações em genes que codificam proteínas essenciais para a liberação de acetilcolina na junção neuromuscular. Os genes associados incluem: MYO9A (miosina não convencional IXa), SLC25A1 (proteína transportadora de tricarboxilato mitocondrial), AGRN (agrina), SNAP25 (proteína 25 associada ao sinaptossoma), SLC5A7 (transportador de colina de alta afinidade 1), CHAT (colina O-acetiltransferase), VAMP1 (proteína 1 associada à membrana vesicular), SYT2 (sinaptotagmina-2), SLC18A3 (transportador vesicular de acetilcolina) e COL13A1 (colágeno alfa-1(XIII)).[1][2][5]
Diagnóstico
O diagnóstico é baseado na avaliação clínica, exames eletrofisiológicos e confirmação genética. A eletromiografia (EMG) pode mostrar anormalidades na transmissão neuromuscular e padrões miopáticos. O sequenciamento completo do exoma (WES) é um dos procedimentos disponíveis para identificar as mutações nos genes associados. Atualmente, há 731 variantes genéticas registradas no ClinVar relacionadas a esta condição.[1][2][5][6]
Tratamento e manejo
O manejo das síndromes miastênicas congênitas pré-sinápticas é multidisciplinar e inclui acompanhamento com neurologista, fisioterapia e terapia ocupacional. O atendimento em reabilitação para doenças raras está disponível no SUS. O tratamento é individualizado e pode incluir medicamentos que melhoram a transmissão neuromuscular, mas a escolha e a dose devem ser definidas pelo médico especialista, com base no tipo de mutação e nos sintomas de cada paciente.[1][2][6]
Tratamentos citados na literatura
A literatura científica (fonte PubTator3) menciona diversos fármacos estudados para estas síndromes, com o seguinte número de publicações associadas: Brometo de Piridostigmina (122 publicações), Albuterol (62), Esteroides (56), Acetilcolina (48), Prednisona (40), Azatioprina (37), Efedrina (35), Neostigmina (31), Prednisolona (30) e Amifampridina (29). É importante destacar que estas são associações mineradas da literatura e não representam uma recomendação de tratamento. A decisão terapêutica deve ser tomada pelo médico assistente.[6]
Prognóstico e qualidade de vida
O prognóstico varia conforme a gravidade dos sintomas e o gene envolvido. Episódios de insuficiência respiratória e apneia súbita podem ser potencialmente graves, exigindo monitoramento contínuo. O acompanhamento regular com uma equipe multidisciplinar e o suporte respiratório adequado são fundamentais para melhorar a qualidade de vida e reduzir complicações.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Síndrome miastênica congênita (SMC) é um distúrbio neuromuscular hereditário causado por defeitos de diversos tipos na junção neuromuscular. Os efeitos da doença são semelhantes aos da síndrome de Lambert-Eaton e da miastenia gravis, sendo a diferença que a SMC não é um distúrbio autoimune. Existem apenas 600 casos familiares conhecidos deste distúrbio e estima-se que sua frequência geral na população humana seja de 1 em 200.000.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
As síndromes miastênicas congênitas pré-sinápticas são um grupo de doenças neuromusculares hereditárias raras que afetam a transmissão do impulso nervoso do nervo para o músculo. O termo 'pré-sináptico' indica que o defeito genético está localizado na parte do neurônio que libera o neurotransmissor (acetilcolina) na junção neuromuscular. Essas condições geralmente se manifestam desde o nascimento (período neonatal) ou na primeira infância, causando fraqueza muscular que piora com o esforço (fadiga).[1][4]
Sinais e sintomas
Os sintomas podem variar de pessoa para pessoa, mas frequentemente incluem fraqueza muscular generalizada, fadiga fácil (fraqueza fatigável) e fraqueza que afeta tanto os músculos próximos ao tronco (proximal) quanto os das extremidades (distal). Muitos pacientes apresentam episódios intermitentes de insuficiência respiratória devido à fraqueza muscular, apneia central do sono, desconforto respiratório episódico e parada respiratória. Outros sinais comuns são arreflexia (ausência de reflexos), polineuropatia motora, olhar fixo, marcha na ponta dos pés, estridor (ruído ao respirar), episódios de engasgos, regurgitação nasal, deformidades da coluna vertebral, rigidez espinhal, atrofia da fibra muscular, amiotrofia distal (perda de massa muscular nas extremidades) e mandíbula estreita.[1][4]
Causas genéticas
Estas síndromes são causadas por mutações em genes que codificam proteínas essenciais para a liberação de acetilcolina na junção neuromuscular. Os genes associados incluem: MYO9A (miosina não convencional IXa), SLC25A1 (proteína transportadora de tricarboxilato mitocondrial), AGRN (agrina), SNAP25 (proteína 25 associada ao sinaptossoma), SLC5A7 (transportador de colina de alta afinidade 1), CHAT (colina O-acetiltransferase), VAMP1 (proteína 1 associada à membrana vesicular), SYT2 (sinaptotagmina-2), SLC18A3 (transportador vesicular de acetilcolina) e COL13A1 (colágeno alfa-1(XIII)).[1][2][5]
Diagnóstico
O diagnóstico é baseado na avaliação clínica, exames eletrofisiológicos e confirmação genética. A eletromiografia (EMG) pode mostrar anormalidades na transmissão neuromuscular e padrões miopáticos. O sequenciamento completo do exoma (WES) é um dos procedimentos disponíveis para identificar as mutações nos genes associados. Atualmente, há 731 variantes genéticas registradas no ClinVar relacionadas a esta condição.[1][2][5][6]
Tratamento e manejo
O manejo das síndromes miastênicas congênitas pré-sinápticas é multidisciplinar e inclui acompanhamento com neurologista, fisioterapia e terapia ocupacional. O atendimento em reabilitação para doenças raras está disponível no SUS. O tratamento é individualizado e pode incluir medicamentos que melhoram a transmissão neuromuscular, mas a escolha e a dose devem ser definidas pelo médico especialista, com base no tipo de mutação e nos sintomas de cada paciente.[1][2][6]
Tratamentos citados na literatura
A literatura científica (fonte PubTator3) menciona diversos fármacos estudados para estas síndromes, com o seguinte número de publicações associadas: Brometo de Piridostigmina (122 publicações), Albuterol (62), Esteroides (56), Acetilcolina (48), Prednisona (40), Azatioprina (37), Efedrina (35), Neostigmina (31), Prednisolona (30) e Amifampridina (29). É importante destacar que estas são associações mineradas da literatura e não representam uma recomendação de tratamento. A decisão terapêutica deve ser tomada pelo médico assistente.[6]
Prognóstico e qualidade de vida
O prognóstico varia conforme a gravidade dos sintomas e o gene envolvido. Episódios de insuficiência respiratória e apneia súbita podem ser potencialmente graves, exigindo monitoramento contínuo. O acompanhamento regular com uma equipe multidisciplinar e o suporte respiratório adequado são fundamentais para melhorar a qualidade de vida e reduzir complicações.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 26 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 67 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
10 genes identificados com associação a esta condição.
Myosins are actin-based motor molecules with ATPase activity. Unconventional myosins serve in intracellular movements. Regulates Rho by stimulating its GTPase activity in neurons. Required for the regulation of neurite branching and motor neuron axon guidance (By similarity)
MembraneCytoplasmSynapseCell projection, growth cone
Myasthenic syndrome, congenital, 24, presynaptic
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features include easy fatigability and muscle weakness. CMS24 inheritance is autosomal recessive.
Mitochondrial electroneutral antiporter that exports citrate from the mitochondria into the cytosol in exchange for malate (PubMed:26870663, PubMed:29031613, PubMed:29238895, PubMed:39881208, PubMed:38937634). Also able to mediate the exchange of citrate for isocitrate, phosphoenolpyruvate, cis-aconitate and to a lesser extent trans-aconitate, maleate and succinate (PubMed:29031613). Substrate exchange across the membrane occurs consecutively with one substrate being transported first, then diss
Mitochondrion inner membrane
Combined D-2- and L-2-hydroxyglutaric aciduria
An autosomal recessive neurometabolic disorder characterized by neonatal-onset encephalopathy with severe muscular weakness, intractable seizures, respiratory distress, and lack of psychomotor development resulting in early death. Brain imaging shows abnormalities including enlarged ventricles, delayed myelination, and germinal layer cysts.
Depending on alternative splicing and post-translational modifications, it has a role in different processes, including neuromuscular junction formation and maintenance, and regulation of neurite outgrowth (By similarity). Also involved in positive regulation of cartilage formation through alpha-dystroglycan binding and up-regulation of SOX9 (PubMed:26290588) Heparan sulfate basal lamina glycoprotein that plays a central role in the formation and the maintenance of the neuromuscular junction (NM
Secreted, extracellular space, extracellular matrixSynapseCell membrane
Myasthenic syndrome, congenital, 8
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS8 is an autosomal recessive disease characterized by prominent defects of both the pre- and postsynaptic regions. Affected individuals have onset of muscle weakness in early childhood; the severity of the weakness and muscles affected is variable.
t-SNARE involved in the molecular regulation of neurotransmitter release. May play an important role in the synaptic function of specific neuronal systems. Associates with proteins involved in vesicle docking and membrane fusion. Regulates plasma membrane recycling through its interaction with CENPF. Modulates the gating characteristics of the delayed rectifier voltage-dependent potassium channel KCNB1 in pancreatic beta cells
Cytoplasm, perinuclear regionCell membraneSynapse, synaptosomePhotoreceptor inner segment
Developmental and epileptic encephalopathy 117
A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE117 is an autosomal dominant form characterized by global developmental delay, delayed walking or inability to walk, intellectual disability, and early-onset seizures. Variable neurological symptoms are muscular hypotonia, movement disorders (ataxia, dystonia or tremor), cerebral visual impairment, and brain volume loss.
High-affinity Na(+)-coupled choline transmembrane symporter (PubMed:11027560, PubMed:11068039, PubMed:12237312, PubMed:12969261, PubMed:17005849, PubMed:23132865, PubMed:23141292, PubMed:27569547). Functions as an electrogenic, voltage-dependent transporter with variable charge/choline stoichiometry (PubMed:17005849). Choline uptake and choline-induced current is also Cl(-)-dependent where Cl(-) is likely a regulatory ion rather than cotransported ion (PubMed:11068039, PubMed:12237312, PubMed:17
Presynaptic cell membraneCell projection, axonEarly endosome membraneCytoplasmic vesicle, secretory vesicle, synaptic vesicle membrane
Neuronopathy, distal hereditary motor, autosomal dominant 7
A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMND7 is characterized by onset in the second decade of progressive distal muscle wasting and weakness affecting the upper and lower limbs and resulting in walking difficulties and hand grip. There is significant muscle atrophy of the hands and lower limbs. The disorder is associated with vocal cord paresis due to involvement of the tenth cranial nerve.
Catalyzes the reversible synthesis of acetylcholine (ACh) from acetyl CoA and choline at cholinergic synapses
Myasthenic syndrome, congenital, 6, presynaptic
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS6 affected individuals have myasthenic symptoms since birth or early infancy, negative tests for anti-AChR antibodies, and abrupt episodic crises with increased weakness, bulbar paralysis, and apnea precipitated by undue exertion, fever, or excitement. CMS6 inheritance is autosomal recessive.
Involved in the targeting and/or fusion of transport vesicles to their target membrane
Cytoplasmic vesicle, secretory vesicle, synaptic vesicle membraneSynapse, synaptosomeCytoplasmic vesicle membraneMitochondrion outer membrane
Spastic ataxia 1, autosomal dominant
An autosomal dominant form of spastic ataxia, a progressive neurodegenerative disorder characterized by lower-limb spasticity and generalized ataxia with dysarthria, impaired ocular movements, and gait disturbance.
Exhibits calcium-dependent phospholipid and inositol polyphosphate binding properties (By similarity). May have a regulatory role in the membrane interactions during trafficking of synaptic vesicles at the active zone of the synapse (By similarity). Plays a role in dendrite formation by melanocytes (PubMed:23999003)
Cytoplasmic vesicle, secretory vesicle, synaptic vesicle membraneCytoplasmic vesicle, secretory vesicle, chromaffin granule membraneCytoplasm
Myasthenic syndrome, congenital, 7A, presynaptic, and distal motor neuropathy, autosomal dominant
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS7A is an autosomal dominant, presynaptic disorder resembling Lambert-Eaton myasthenic syndrome. Affected individuals have a variable degree of proximal and distal limb weakness, muscle fatigue that improves with rest, mild gait difficulties, and reduced or absent deep tendon reflexes.
Electrogenic antiporter that exchanges one cholinergic neurotransmitter, acetylcholine or choline, with two intravesicular protons across the membrane of synaptic vesicles. Uses the electrochemical proton gradient established by the V-type proton-pump ATPase to store neurotransmitters inside the vesicles prior to their release via exocytosis (By similarity) (PubMed:20225888, PubMed:8910293). Determines cholinergic vesicular quantal size at presynaptic nerve terminals in developing neuro-muscular
Cytoplasmic vesicle, secretory vesicle, synaptic vesicle membrane
Myasthenic syndrome, congenital, 21, presynaptic
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS21 is an autosomal recessive, pre-synaptic form characterized by ptosis, ophthalmoplegia, fatigable weakness, apneic crises, and deterioration of symptoms in cold water. Learning difficulties and left ventricular dysfunction may be present in some patients.
Involved in cell-matrix and cell-cell adhesion interactions that are required for normal development. May participate in the linkage between muscle fiber and basement membrane. May play a role in endochondral ossification of bone and branching morphogenesis of lung. Binds heparin. At neuromuscular junctions, may play a role in acetylcholine receptor clustering (PubMed:26626625)
Cell membranePostsynaptic cell membrane
Myasthenic syndrome, congenital, 19
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort.
Variantes genéticas (ClinVar)
731 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
50 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndromes miastênicas, congênitas, pré-sinápticas
Centros de Referência SUS
24 centros habilitados pelo SUS para Síndromes miastênicas, congênitas, pré-sinápticas
Centros para Síndromes miastênicas, congênitas, pré-sinápticas
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Presynaptic Congenital Myasthenic Syndromes: Understanding Clinical Phenotypes through In vivo Models.
Presynaptic congenital myasthenic syndromes (CMS) are a group of genetic disorders affecting the presynaptic side of the neuromuscular junctions (NMJ). They can result from a dysfunction in acetylcholine (ACh) synthesis or recycling, in its packaging into synaptic vesicles, or its subsequent release into the synaptic cleft. Other proteins involved in presynaptic endplate development and maintenance can also be impaired.Presynaptic CMS usually presents during the prenatal or neonatal period, with a severe phenotype including congenital arthrogryposis, developmental delay, and apnoeic crisis. However, milder phenotypes with proximal muscle weakness and good response to treatment have been described. Finally, many presynaptic genes are expressed in the brain, justifying the presence of additional central nervous system symptoms.Several animal models have been developed to study CMS, providing the opportunity to identify disease mechanisms and test treatment options. In this review, we describe presynaptic CMS phenotypes with a focus on in vivo models, to better understand CMS pathophysiology and define new causative genes.
New recessive mutations in SYT2 causing severe presynaptic congenital myasthenic syndromes.
To report the identification of 2 new homozygous recessive mutations in the synaptotagmin 2 (SYT2) gene as the genetic cause of severe and early presynaptic forms of congenital myasthenic syndromes (CMSs). Next-generation sequencing identified new homozygous intronic and frameshift mutations in the SYT2 gene as a likely cause of presynaptic CMS. We describe the clinical and electromyographic patient phenotypes, perform ex vivo splicing analyses to characterize the effect of the intronic mutation on exon splicing, and analyze the functional impact of this variation at the neuromuscular junction (NMJ). The 2 infants presented a similar clinical phenotype evoking first a congenital myopathy characterized by muscle weakness and hypotonia. Next-generation sequencing allowed to the identification of 1 homozygous intronic mutation c.465+1G>A in patient 1 and another homozygous frameshift mutation c.328_331dup in patient 2, located respectively in the 5' splice donor site of SYT2 intron 4 and in exon 3. Functional studies of the intronic mutation validated the abolition of the splice donor site of exon 4 leading to its skipping. In-frame skipping of exon 4 that encodes part of the C2A calcium-binding domain of SYT2 is associated with a loss-of-function effect resulting in a decrease of neurotransmitter release and severe pre- and postsynaptic NMJ defects. This study identifies new homozygous recessive SYT2 mutations as the underlying cause of severe and early presynaptic form of CMS expanding the genetic spectrum of recessive SYT2-related CMS associated with defects in neurotransmitter release.
The Electrophysiology of Presynaptic Congenital Myasthenic Syndromes With and Without Facilitation: From Electrodiagnostic Findings to Molecular Mechanisms.
Congenital myasthenic syndromes (CMS) are a group of inherited disorders of neuromuscular transmission most commonly presenting with early onset fatigable weakness, ptosis, and ophthalmoparesis. CMS are classified according to the localization of the causative molecular defect. CMS with presynaptic dysfunction can be caused by mutations in several different genes, including those involved in acetylcholine synthesis, its packaging into synaptic vesicles, vesicle docking, and release from the presynaptic nerve terminal and neuromuscular junction development and maintenance. Electrodiagnostic testing is key in distinguishing CMS from other neuromuscular disorders with similar clinical features as well as for revealing features pointing to a specific molecular diagnosis. A decremental response on low-frequency repetitive nerve stimulation (RNS) is present in most presynaptic CMS. In CMS with deficits in acetylcholine resynthesis however, a decrement may only appear after conditioning with exercise or high-frequency RNS and characteristically displays a slow recovery. Facilitation occurs in CMS caused by mutations in VAMP1, UNC13A, SYT2, AGRN, LAMA5. By contrast, facilitation is absent in the other presynaptic CMS described to date. An understanding of the underlying molecular mechanisms therefore assists the interpretation of electrodiagnostic findings in patients with suspected CMS.
Publicações recentes
Presynaptic Congenital Myasthenic Syndromes: Understanding Clinical Phenotypes through In vivo Models.
New recessive mutations in SYT2 causing severe presynaptic congenital myasthenic syndromes.
The Electrophysiology of Presynaptic Congenital Myasthenic Syndromes With and Without Facilitation: From Electrodiagnostic Findings to Molecular Mechanisms.
Synaptotagmin 2 mutations cause an autosomal-dominant form of lambert-eaton myasthenic syndrome and nonprogressive motor neuropathy.
📚 EuropePMC3 artigos no totalmostrando 3
Presynaptic Congenital Myasthenic Syndromes: Understanding Clinical Phenotypes through In vivo Models.
Journal of neuromuscular diseasesNew recessive mutations in SYT2 causing severe presynaptic congenital myasthenic syndromes.
Neurology. GeneticsThe Electrophysiology of Presynaptic Congenital Myasthenic Syndromes With and Without Facilitation: From Electrodiagnostic Findings to Molecular Mechanisms.
Frontiers in neurologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndromes miastênicas, congênitas, pré-sinápticas.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndromes miastênicas, congênitas, pré-sinápticas
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Presynaptic Congenital Myasthenic Syndromes: Understanding Clinical Phenotypes through In vivo Models.
- New recessive mutations in SYT2 causing severe presynaptic congenital myasthenic syndromes.
- The Electrophysiology of Presynaptic Congenital Myasthenic Syndromes With and Without Facilitation: From Electrodiagnostic Findings to Molecular Mechanisms.
- Synaptotagmin 2 mutations cause an autosomal-dominant form of lambert-eaton myasthenic syndrome and nonprogressive motor neuropathy.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:98914(Orphanet)
- MONDO:0700466(MONDO)
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q56014417(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndromes miastênicas, congênitas, pré-sinápticas
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- Dado público estruturado
- fonte: Wikidata