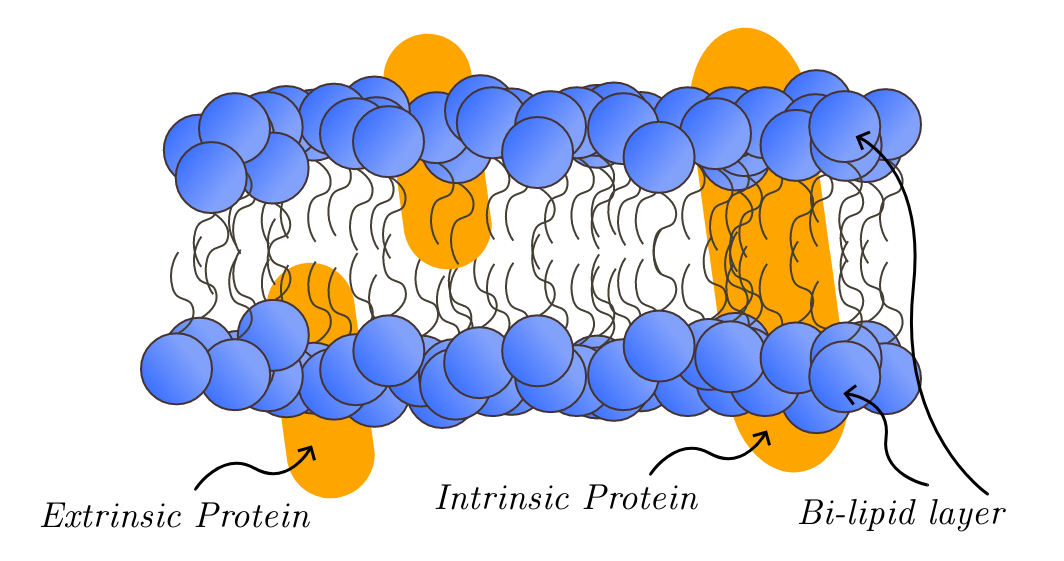
O complexo oligomérico de Golgi conservado, também conhecido como complexo COG, é uma proteína de membrana extrínseca encontrada no aparelho de Golgi em eucariotos. Este complexo é conservado evolutivamente e possui subunidades homólogas encontradas em todas as espécies de eucariotos. O complexo COG foi descoberto pela primeira vez em 1981 em células de ovário de hamster chinês. Essas células de ovário apresentavam mutações nos receptores de lipoproteína de baixa densidade (LDL), o que afetava a função das enzimas de glicosilação do Golgi. No entanto, o complexo COG como um todo não foi totalmente compreendido até 2004, quando uma deficiência em uma das subunidades do COG foi associada a distúrbios congênitos da glicosilação. Essas descobertas levaram à percepção de que o complexo COG desempenha um papel importante na glicosilação e na classificação de proteínas.
Introdução
O que você precisa saber de cara
Visão geral
A COG2-CDG é uma doença genética rara que faz parte do grupo das doenças congênitas da glicosilação (CDG). A glicosilação é um processo essencial para o funcionamento das células, e sua alteração pode afetar diversos órgãos e sistemas. A condição é herdada de forma autossômica recessiva, ou seja, é necessário herdar uma cópia do gene alterado de cada um dos pais para desenvolver a doença. Estima-se que sua prevalência seja inferior a 1 caso a cada 1.000.000 de pessoas, e os primeiros sinais geralmente aparecem na infância.[1][4]
Sinais e sintomas
Os sintomas da COG2-CDG são variados e podem afetar múltiplos sistemas do corpo. Entre os sinais mais comuns estão: atraso global do desenvolvimento, deficiência intelectual, hipotonia (tônus muscular diminuído) e microcefalia secundária (cabeça menor que o esperado, que se desenvolve após o nascimento). Também podem ocorrer tetraplegia espástica (paralisia dos quatro membros com rigidez muscular), crises tônicas generalizadas (um tipo de convulsão) e deterioração psicomotora (perda de habilidades já adquiridas).[1][4]
Sinais e sintomas (continuação)
Alterações neurológicas estruturais são frequentes, como atrofia cerebral difusa (perda de tecido cerebral em várias áreas), hipoplasia do corpo caloso (desenvolvimento incompleto da estrutura que conecta os dois hemisférios cerebrais) e glândula pituitária pequena. Além disso, podem ser observadas anormalidades no formato do rosto, comprometimento da função hepática (com elevação das transaminases hepáticas no sangue), alterações na cascata de coagulação e níveis diminuídos de cobre e ceruloplasmina circulantes. A glicosilação anormal é uma característica laboratorial marcante da doença.[1][4]
Causas genéticas
A COG2-CDG é causada por alterações (mutações) no gene COG2, que fornece instruções para a produção da subunidade 2 do complexo oligomérico conservado do Golgi (COG). Esse complexo é fundamental para o funcionamento adequado do aparelho de Golgi, uma organela celular responsável pela modificação e transporte de proteínas. Mutações nesse gene prejudicam a glicosilação, levando aos sintomas da doença. A herança é autossômica recessiva, e o início dos sintomas ocorre na infância.[1][2][5]
Diagnóstico
O diagnóstico da COG2-CDG é baseado na combinação de achados clínicos, exames laboratoriais e testes genéticos. Exames de sangue podem mostrar alterações como níveis elevados de transaminases hepáticas, diminuição de cobre e ceruloplasmina, e anormalidades na coagulação. A análise da glicosilação de proteínas séricas (como a transferrina) é um exame importante para identificar o padrão característico de glicosilação anormal. O diagnóstico definitivo é confirmado pelo sequenciamento completo do exoma (WES) ou por painel genético que identifique mutações no gene COG2. Atualmente, há 54 variantes reportadas no ClinVar e 8 testes genéticos disponíveis para a condição.[1][2][5]
Diagnóstico (SUS)
No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para o diagnóstico da COG2-CDG. Entre os procedimentos disponíveis estão: teste do pezinho (triagem neonatal), dosagem de aminoácidos (para erros inatos do metabolismo), dosagem de ácidos orgânicos na urina, teste de triagem para erros inatos do metabolismo e sequenciamento completo do exoma (WES). O atendimento em reabilitação para doenças raras também é um recurso disponível.[1][2]
Tratamento e manejo
Atualmente, não há cura para a COG2-CDG, e o tratamento é focado no manejo dos sintomas e na melhora da qualidade de vida. A abordagem é multidisciplinar e pode incluir: acompanhamento neurológico para controle de crises convulsivas e espasticidade, suporte nutricional, fisioterapia, terapia ocupacional e fonoaudiologia. O monitoramento da função hepática e dos níveis de cobre e ceruloplasmina também é importante. Não há medicamentos específicos aprovados para a doença, e nenhum fármaco foi listado como tratamento na literatura científica revisada até o momento.[1][2]
Prognóstico e qualidade de vida
O prognóstico da COG2-CDG varia de acordo com a gravidade dos sintomas e a rapidez do diagnóstico e intervenção. Por ser uma doença de início na infância, o acompanhamento precoce com uma equipe multidisciplinar é essencial para maximizar o desenvolvimento e a qualidade de vida. O suporte familiar e o acesso a serviços de reabilitação são fundamentais para lidar com os desafios motores, intelectuais e clínicos ao longo da vida.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
O complexo oligomérico de Golgi conservado, também conhecido como complexo COG, é uma proteína de membrana extrínseca encontrada no aparelho de Golgi em eucariotos. Este complexo é conservado evolutivamente e possui subunidades homólogas encontradas em todas as espécies de eucariotos. O complexo COG foi descoberto pela primeira vez em 1981 em células de ovário de hamster chinês. Essas células de ovário apresentavam mutações nos receptores de lipoproteína de baixa densidade (LDL), o que afetava a função das enzimas de glicosilação do Golgi. No entanto, o complexo COG como um todo não foi totalmente compreendido até 2004, quando uma deficiência em uma das subunidades do COG foi associada a distúrbios congênitos da glicosilação. Essas descobertas levaram à percepção de que o complexo COG desempenha um papel importante na glicosilação e na classificação de proteínas.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A COG2-CDG é uma doença genética rara que faz parte do grupo das doenças congênitas da glicosilação (CDG). A glicosilação é um processo essencial para o funcionamento das células, e sua alteração pode afetar diversos órgãos e sistemas. A condição é herdada de forma autossômica recessiva, ou seja, é necessário herdar uma cópia do gene alterado de cada um dos pais para desenvolver a doença. Estima-se que sua prevalência seja inferior a 1 caso a cada 1.000.000 de pessoas, e os primeiros sinais geralmente aparecem na infância.[1][4]
Sinais e sintomas
Os sintomas da COG2-CDG são variados e podem afetar múltiplos sistemas do corpo. Entre os sinais mais comuns estão: atraso global do desenvolvimento, deficiência intelectual, hipotonia (tônus muscular diminuído) e microcefalia secundária (cabeça menor que o esperado, que se desenvolve após o nascimento). Também podem ocorrer tetraplegia espástica (paralisia dos quatro membros com rigidez muscular), crises tônicas generalizadas (um tipo de convulsão) e deterioração psicomotora (perda de habilidades já adquiridas).[1][4]
Sinais e sintomas (continuação)
Alterações neurológicas estruturais são frequentes, como atrofia cerebral difusa (perda de tecido cerebral em várias áreas), hipoplasia do corpo caloso (desenvolvimento incompleto da estrutura que conecta os dois hemisférios cerebrais) e glândula pituitária pequena. Além disso, podem ser observadas anormalidades no formato do rosto, comprometimento da função hepática (com elevação das transaminases hepáticas no sangue), alterações na cascata de coagulação e níveis diminuídos de cobre e ceruloplasmina circulantes. A glicosilação anormal é uma característica laboratorial marcante da doença.[1][4]
Causas genéticas
A COG2-CDG é causada por alterações (mutações) no gene COG2, que fornece instruções para a produção da subunidade 2 do complexo oligomérico conservado do Golgi (COG). Esse complexo é fundamental para o funcionamento adequado do aparelho de Golgi, uma organela celular responsável pela modificação e transporte de proteínas. Mutações nesse gene prejudicam a glicosilação, levando aos sintomas da doença. A herança é autossômica recessiva, e o início dos sintomas ocorre na infância.[1][2][5]
Diagnóstico
O diagnóstico da COG2-CDG é baseado na combinação de achados clínicos, exames laboratoriais e testes genéticos. Exames de sangue podem mostrar alterações como níveis elevados de transaminases hepáticas, diminuição de cobre e ceruloplasmina, e anormalidades na coagulação. A análise da glicosilação de proteínas séricas (como a transferrina) é um exame importante para identificar o padrão característico de glicosilação anormal. O diagnóstico definitivo é confirmado pelo sequenciamento completo do exoma (WES) ou por painel genético que identifique mutações no gene COG2. Atualmente, há 54 variantes reportadas no ClinVar e 8 testes genéticos disponíveis para a condição.[1][2][5]
Diagnóstico (SUS)
No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para o diagnóstico da COG2-CDG. Entre os procedimentos disponíveis estão: teste do pezinho (triagem neonatal), dosagem de aminoácidos (para erros inatos do metabolismo), dosagem de ácidos orgânicos na urina, teste de triagem para erros inatos do metabolismo e sequenciamento completo do exoma (WES). O atendimento em reabilitação para doenças raras também é um recurso disponível.[1][2]
Tratamento e manejo
Atualmente, não há cura para a COG2-CDG, e o tratamento é focado no manejo dos sintomas e na melhora da qualidade de vida. A abordagem é multidisciplinar e pode incluir: acompanhamento neurológico para controle de crises convulsivas e espasticidade, suporte nutricional, fisioterapia, terapia ocupacional e fonoaudiologia. O monitoramento da função hepática e dos níveis de cobre e ceruloplasmina também é importante. Não há medicamentos específicos aprovados para a doença, e nenhum fármaco foi listado como tratamento na literatura científica revisada até o momento.[1][2]
Prognóstico e qualidade de vida
O prognóstico da COG2-CDG varia de acordo com a gravidade dos sintomas e a rapidez do diagnóstico e intervenção. Por ser uma doença de início na infância, o acompanhamento precoce com uma equipe multidisciplinar é essencial para maximizar o desenvolvimento e a qualidade de vida. O suporte familiar e o acesso a serviços de reabilitação são fundamentais para lidar com os desafios motores, intelectuais e clínicos ao longo da vida.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 6 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 19 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisRequired for normal Golgi morphology and function
Golgi apparatus membrane
Congenital disorder of glycosylation 2Q
A form of congenital disorder of glycosylation, a genetically heterogeneous group of autosomal recessive, multisystem disorders caused by a defect in glycoprotein biosynthesis and characterized by under-glycosylated serum glycoproteins. Congenital disorders of glycosylation result in a wide variety of clinical features, such as defects in the nervous system development, psychomotor retardation, dysmorphic features, hypotonia, coagulation disorders, and immunodeficiency. The broad spectrum of features reflects the critical role of N-glycoproteins during embryonic development, differentiation, and maintenance of cell functions. The transmission pattern of CDG2Q is consistent with autosomal recessive inheritance.
Variantes genéticas (ClinVar)
54 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
3 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — COG2-CDG
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Publicações recentes
Mast cell mediators in hereditary angioedema.
Prenatal Molecular Diagnosis of COL2A1-Associated Stickler Syndrome: Genotype-Phenotype Correlation in a Resource-Limited Healthcare Setting.
Platelet gene signatures detecting pulmonary artery stenosis in patients with pulmonary hypertension.
The global impact of imiglucerase therapy in children with Gaucher disease types 1 and 3: a real-world analysis from the International Collaborative Gaucher Group Gaucher Registry.
Monogenic lupus with SLC7A7 mutations: a retrospective study from a Chinese center.
Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para COG2-CDG.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para COG2-CDG
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Mast cell mediators in hereditary angioedema.
- Prenatal Molecular Diagnosis of COL2A1-Associated Stickler Syndrome: Genotype-Phenotype Correlation in a Resource-Limited Healthcare Setting.
- Platelet gene signatures detecting pulmonary artery stenosis in patients with pulmonary hypertension.
- The global impact of imiglucerase therapy in children with Gaucher disease types 1 and 3: a real-world analysis from the International Collaborative Gaucher Group Gaucher Registry.
- Monogenic lupus with SLC7A7 mutations: a retrospective study from a Chinese center.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:435934(Orphanet)
- OMIM OMIM:617395(OMIM)
- MONDO:0054559(MONDO)
- GARD:17720(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q60195127(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
COG2-CDG
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata