A deficiência de carnitina palmitoiltransferase II (CPT II) na sua forma neonatal é uma doença hereditária que impede a queima adequada de certos tipos de gordura (chamados ácidos graxos de cadeia longa, ou AGCL) nas mitocôndrias, que são as "usinas de energia" das nossas células. Esta é a versão mais grave e fatal da doença, que leva à falha de múltiplos órgãos do corpo.
Introdução
O que você precisa saber de cara
A deficiência de carnitina palmitoiltransferase II (CPT II) na sua forma neonatal é uma doença hereditária que impede a queima adequada de certos tipos de gordura (chamados ácidos graxos de cadeia longa, ou AGCL) nas mitocôndrias, que são as "usinas de energia" das nossas células. Esta é a versão mais grave e fatal da doença, que leva à falha de múltiplos órgãos do corpo.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 31 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 98 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisInvolved in the intramitochondrial synthesis of acylcarnitines from accumulated acyl-CoA metabolites (PubMed:20538056, PubMed:24780397). Reconverts acylcarnitines back into the respective acyl-CoA esters that can then undergo beta-oxidation, an essential step for the mitochondrial uptake of long-chain fatty acids and their subsequent beta-oxidation in the mitochondrion. Active with medium (C8-C12) and long-chain (C14-C18) acyl-CoA esters (PubMed:20538056)
Mitochondrion inner membrane
Carnitine palmitoyltransferase 2 deficiency, myopathic, stress-induced
An autosomal recessive disorder of mitochondrial long-chain fatty acid oxidation, characterized by recurrent myoglobinuria, episodes of muscle pain, stiffness, and rhabdomyolysis. These symptoms are exacerbated by prolonged exercise, fasting, cold, or viral infection. CPT2DM affects most frequently children or young adults, and severity of attacks is highly variable. Myoglobinuria can cause kidney failure and death.
Variantes genéticas (ClinVar)
308 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 235 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Deficiência da carnitina-palmitoil transferase 2, forma neonatal
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Carnitine palmitoyltransferase II (CPT II) deficiency responsible for refractory cardiac arrhythmias, acute multiorgan failure and early fatal outcome.
Carnitine palmitoyltransferase II (CPT II) deficiency is a rare inborn error of mitochondrial fatty acid metabolism with autosomal recessive pattern of inheritance. Its phenotype is highly variable (neonatal, infantile, and adult onset) on the base of mutations of the CPT II gene. In affected subjects, long-chain acylcarnitines cannot be subdivided into carnitine and acyl-CoA, leading to their toxic accumulation in different organs. Neonatal form is the most severe, and all the reported patients died within a few days to 6 months after birth. Hereby, we report on a male late-preterm newborn who presented refractory cardiac arrhythmias and acute multiorgan (hepatic, renal, muscular) injury, leading to cerebral hemorrhage, hydrocephalus, cardiovascular failure and early (day 5 of life) to death. Subsequently, extended metabolic screening and target next generation sequencing (NGS) analysis allowed the CPT II deficiency diagnosis. The male proband was born at 36+ 4 weeks of gestation by spontaneous vaginal delivery. Parents were healthy and nonconsanguineous, although both coming from Nigeria. Family history was unremarkable. Apgar score was 9/9. At birth, anthropometric measures were as follows: weight 2850 g (47th centile, -0.07 standard deviations, SD), length 50 cm (81st centile, + 0.89 SD) and occipitofrontal circumference (OFC) 35 cm (87th centile, + 1.14 SD). On day 2 of life our newborn showed bradycardia (heart rate around 80 bpm) and hypotonia, and was then transferred to the Neonatal Intensive Care Unit (NICU). There, he subsequently manifested many episodes of ventricular tachycardia, which were treated with pharmacological (magnesium sulfate) and electrical cardioversion. Due to the critical conditions of the baby (hepatic, renal and cardiac dysfunctions) and to guarantee optimal management of the arrythmias, he was transferred to the Pediatric Cardiology Reference Center of our region (Sicily, Italy), where he died 2 days later. Thereafter, the carnitines profile evidenced by the extended metabolic screening resulted compatible with a fatty acid oxidation defect (increased levels of acylcarnitines C16 and C18, and low of C2); afterwards, the targeted next generation sequencing (NGS) analysis revealed the known c.680 C > T p. (Pro227Leu) homozygous missense mutation of the CPTII gene, for diagnosis of CPT II deficiency. Genetic investigations have been, then, extended to the baby's parents, who were identified as heterozygous carriers of the same variant. When we meet again the parents for genetic counseling, the mother was within the first trimester of her second pregnancy. Therefore, we offered to the couple and performed the prenatal target NGS analysis on chorionic villi sample, which did not detect any alterations, excluding thus the CPT II deficiency in their second child. CPTII deficiency may be suspected in newborns showing cardiac arrhythmias, associated or not with hypertrophic cardiomyopathy, polycystic kidneys, brain malformations, hepatomegaly. Its diagnosis should be even more suspected and investigated in cases of increased plasmatic levels of creatine phosphokinase and acylcarnitines in addition to kidney, heart and liver dysfunctions, as occurred in the present patient. Accurate family history, extended metabolic screening, and multidisciplinary approach are necessary for diagnosis and adequate management of affected subjects. Next generation sequencing (NGS) techniques allow the identification of the CPTII gene mutation, essential to confirm the diagnosis before or after birth, as well as to calculate the recurrence risk for family members. Our report broads the knowledge of the genetic and molecular bases of such rare disease, improving its clinical characterization, and provides useful indications for the treatment of patients.
Experience with carnitine palmitoyltransferase II deficiency: diagnostic challenges in the myopathic form.
Carnitine palmitoyltransferase II (CPT II) deficiency is an autosomal recessive disorder of long-chain fatty acid oxidation. Three clinical phenotypes, lethal neonatal form, severe infantile hepatocardiomuscular form, and myopathic form, have been described in CPT II deficiency. The myopathic form is usually mild and can manifest from infancy to adulthood, characterised by recurrent rhabdomyolysis episodes. The study aimed to investigate the clinical features, biochemical, histopathological, and genetic findings of 13 patients diagnosed with the myopathic form of CPT II deficiency at Ege University Hospital. A retrospective study was conducted with 13 patients with the myopathic form of CPT II deficiency. Our study considered demographic data, triggers of recurrent rhabdomyolysis attacks, biochemical metabolic screening, and molecular analysis. Ten patients were examined for rhabdomyolysis of unknown causes. Two patients were diagnosed during family screening, and one was diagnosed during investigations due to increased liver function tests. Acylcarnitine profiles were normal in five patients during rhabdomyolysis. Genetic studies have identified a c.338C>T (p.Ser113Leu) variant homozygous in 10 patients. One patient showed a novel frameshift variant compound heterozygous with c.338C>T (p.Ser113Leu). Plasma acylcarnitine analysis should be preferred as it is superior to DBS acylcarnitine analysis in diagnosing CPT II deficiency. Even if plasma acylcarnitine analysis is impossible, CPT2 gene analysis should be performed. Our study emphasizes that CPT II deficiency should be considered in the differential diagnosis of recurrent rhabdomyolysis, even if typical acylcarnitine elevation does not accompany it.
Do renal and cardiac malformations in the fetus signal carnitine palmitoyltransferase II deficiency? A rare lethal fatty acid oxidation defect.
The neonatal form of carnitine palmitoyltransferase II (CPT II) deficiency is a rare lethal inherited disorder of fatty acid oxidation. Carnitine essentially transfers long-chain fatty acids across the mitochondrial membranes for β-oxidation, where CPT II plays a key role. CPT II deficiency phenotypical forms include lethal neonatal, severe infantile and myopathic forms. We present a term small-for-gestational-age neonate with hypoglycaemia, seizures, refractory cardiac arrhythmias and intracranial haemorrhage. Plasma acylcarnitine profile and the genetic study confirmed CPT II deficiency. Additionally, likely pathogenic variants in the SLC22A5 gene point to primary carnitine deficiency. Antenatal findings of polycystic kidney disease and cardiomegaly were confirmed postnatally. All supportive measures, including extracorporeal life support, failed to improve the clinical course, and the baby succumbed. Major renal, cerebral and cardiac anomalies were reported with CPT II deficiency. In our case, fetal polycystic nephromegaly and cardiomegaly with parental consanguinity should have signalled the possibility of this disorder.
Cause of recurrent rhabdomyolysis, carnitine palmitoyltransferase II deficiency and novel pathogenic mutation.
Carnitine palmitoyltransferase II (CPT II) deficiency is an autosomal inherited metabolic disorder in which the β-oxidation of the long chain fatty acids is defective. The clinical presentation may be in various forms; it presents itself in the severe form during neonatal and infantile periods and as the less severe myopathic form in the school age and adolescence. While the severity of the rhabdomyolysis attacks varies, occasionally the clinical course may be complicated with acute renal failure. Acylcarnitine analysis may help in the diagnosis of CPT II, but its normality does not indicate the absence of the disease. If there is strong suspicion, genetic analysis should be performed on the cases. In this article, we present a 15-year-old male patient who had two rhabdomyolysis attacks triggered by infection and starvation. Acylcarnitine analysis of the case was normal, CPT II deficiency was considered when the history was evaluated, and CPT II gene c.137A>G (p.Gln46Arg) homozygous novel pathogenic mutation was detected. CPT II deficiency is one of the most common causes of metabolic rhabdomyolysis in patients with recurrent episodes of rhabdomyolysis. A karnitin-palmitoil-transzferáz II- (CPT II-) hiány egy autoszomálisan öröklődő anyagcsere-rendellenesség, amelyben a hosszú láncú zsírsavak β-oxidációja hiányos. A klinikai megjelenés különféle formában lehetséges; súlyos formájában jelentkezik újszülöttkorban és infantilis időszakban, míg iskolás- és serdülőkorban a kevésbé súlyos myopathiás formában jelentkezik. Bár a rhabdomyolysisrohamok súlyossága változó, a klinikai lefolyást esetenként akut veseelégtelenség komplikálhatja. Az acilkarnitin-elemzés segíthet a CPT II diagnosztizálásában, de az eredmény normalitása nem jelzi a betegség hiányát. Erős gyanú esetén genetikai elemzést kell végezni. Ebben a tanulmányban egy 15 éves fiú beteg esetét mutatjuk be, akinek két, fertőzés, illetve éhezés által kiváltott rhabdomyolysisrohama volt. Az acilkarnitin-elemzés eredménye normális volt, a kórtörténet értékelésénél figyelembe vettük a CPT II-hiányt, és kimutattuk a CPT II gén c.137A> G (p.Gln46Arg) új patogén homozigóta mutációját. A CPT II-hiány a metabolikus rhabdomyolysis egyik leggyakoribb oka az ismétlődő rhabdomyolysisepizódoktól szenvedő betegek esetén.
Muscle Carnitine Palmitoyltransferase II (CPT II) Deficiency: A Conceptual Approach.
Carnitine palmitoyltransferase (CPT) catalyzes the transfer of long- and medium-chain fatty acids from cytoplasm into mitochondria, where oxidation of fatty acids takes place. Deficiency of CPT enzyme is associated with rare diseases of fatty acid metabolism. CPT is present in two subforms: CPT I at the outer mitochondrial membrane and carnitine palmitoyltransferase II (CPT II) inside the mitochondria. Deficiency of CPT II results in the most common inherited disorder of long-chain fatty acid oxidation affecting skeletal muscle. There is a lethal neonatal form, a severe infantile hepato-cardio-muscular form, and a rather mild myopathic form characterized by exercise-induced myalgia, weakness, and myoglobinuria. Total CPT activity (CPT I + CPT II) in muscles of CPT II-deficient patients is generally normal. Nevertheless, in some patients, not detectable to reduced total activities are also reported. CPT II protein is also shown in normal concentration in patients with normal CPT enzymatic activity. However, residual CPT II shows abnormal inhibition sensitivity towards malonyl-CoA, Triton X-100 and fatty acid metabolites in patients. Genetic studies have identified a common p.Ser113Leu mutation in the muscle form along with around 100 different rare mutations. The biochemical consequences of these mutations have been controversial. Hypotheses include lack of enzymatically active protein, partial enzyme deficiency and abnormally regulated enzyme. The recombinant enzyme experiments that we recently conducted have shown that CPT II enzyme is extremely thermoliable and is abnormally inhibited by different emulsifiers and detergents such as malonyl-CoA, palmitoyl-CoA, palmitoylcarnitine, Tween 20 and Triton X-100. Here, we present a conceptual overview on CPT II deficiency based on our own findings and on results from other studies addressing clinical, biochemical, histological, immunohistological and genetic aspects, as well as recent advancements in diagnosis and therapeutic strategies in this disorder.
Publicações recentes
Carnitine palmitoyltransferase II (CPT II) deficiency responsible for refractory cardiac arrhythmias, acute multiorgan failure and early fatal outcome.
Do renal and cardiac malformations in the fetus signal carnitine palmitoyltransferase II deficiency? A rare lethal fatty acid oxidation defect.
Successful orthotopic heart transplantation in CPTII deficiency.
[Tandem mass spectrometry analysis and genetic diagnosis of neonates with fatty acid oxidation disorders in central and northern regions of Guangxi].
Epidemiology of rare diseases detected by newborn screening in the Czech Republic.
📚 EuropePMCmostrando 7
Carnitine palmitoyltransferase II (CPT II) deficiency responsible for refractory cardiac arrhythmias, acute multiorgan failure and early fatal outcome.
Italian journal of pediatricsExperience with carnitine palmitoyltransferase II deficiency: diagnostic challenges in the myopathic form.
Journal of pediatric endocrinology & metabolism : JPEMDo renal and cardiac malformations in the fetus signal carnitine palmitoyltransferase II deficiency? A rare lethal fatty acid oxidation defect.
BMJ case reportsCause of recurrent rhabdomyolysis, carnitine palmitoyltransferase II deficiency and novel pathogenic mutation.
Ideggyogyaszati szemleMuscle Carnitine Palmitoyltransferase II (CPT II) Deficiency: A Conceptual Approach.
Molecules (Basel, Switzerland)Carnitine palmitoyltransferase II deficiency with a focus on newborn screening.
Journal of human geneticsAcute Respiratory Infection Unveiling CPT II Deficiency.
International journal of molecular sciencesAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Deficiência da carnitina-palmitoil transferase 2, forma neonatal.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Deficiência da carnitina-palmitoil transferase 2, forma neonatal
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Carnitine palmitoyltransferase II (CPT II) deficiency responsible for refractory cardiac arrhythmias, acute multiorgan failure and early fatal outcome.
- Experience with carnitine palmitoyltransferase II deficiency: diagnostic challenges in the myopathic form.
- Do renal and cardiac malformations in the fetus signal carnitine palmitoyltransferase II deficiency? A rare lethal fatty acid oxidation defect.
- Cause of recurrent rhabdomyolysis, carnitine palmitoyltransferase II deficiency and novel pathogenic mutation.
- Muscle Carnitine Palmitoyltransferase II (CPT II) Deficiency: A Conceptual Approach.
- Successful orthotopic heart transplantation in CPTII deficiency.
- [Tandem mass spectrometry analysis and genetic diagnosis of neonates with fatty acid oxidation disorders in central and northern regions of Guangxi].
- Epidemiology of rare diseases detected by newborn screening in the Czech Republic.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:228308(Orphanet)
- OMIM OMIM:608836(OMIM)
- MONDO:0012136(MONDO)
- GARD:17151(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
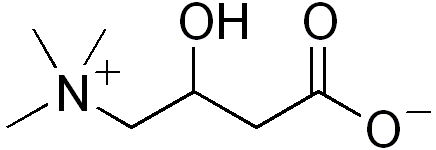
Deficiência da carnitina-palmitoil transferase 2, forma neonatal
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)