Inflamação da hipófise causada pelo próprio sistema de defesa do corpo, muitas vezes associada a outras doenças autoimunes (como a doença de Hashimoto, a doença de Graves e a doença de Addison).
Introdução
O que você precisa saber de cara
Inflamação da hipófise causada pelo próprio sistema de defesa do corpo, muitas vezes associada a outras doenças autoimunes (como a doença de Hashimoto, a doença de Graves e a doença de Addison).
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 18 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 35 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Deficiência da hormona pituitária de origem autoimune
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Primary hypophysitis: Classification review.
Primary (idiopathic) hypophysitis is traditionally classified into lymphocytic, granulomatous, necrotizing, or IgG4-related disease types. Secondary hypophysitis occurs in patients with systemic conditions, which if known, often obviate the need for biopsy. Primary idiopathic hypophysitis, in contrast, often mimics tumors and mandates histological confirmation of inflammation. We detail four primary hypophysitis cases, discussing challenges in histological classification. Four women, ages 32 to 76 years, presented with weakness, visual changes, fatigue, weight loss, and/or headache. Preoperatively, pituitary macroadenoma/pituitary neuroendocrine tumor was suspected. Biopsies revealed lymphocytic hypophysitis without (Case 1) and with (Case 2) IgG4+ cells (modest numbers) and granulomatous hypophysitis with necrosis, large numbers of IgG4+ cells (Cases 3 and 4), with multinucleated giant cells (Case 4). Steroid therapy, in one case with rituximab, was administered irrespective of histological classification. Lymphocytic hypophysitis comes to biopsy primarily in patients without known risk factors, eg pregnancy or drug usage. Granulomatous or necrotizing types are less common, less histologically uniform, and may contain large numbers of IgG4+ cells, a feature found both in IgG4-related disease and autoimmune disorders, especially granulomatosis with polyangiitis. In certain cases, the use of steroids, followed by rituximab for all types of hypophysitis based on clinical criteria may obviate the need for precise histological distinction.
An update on hypophysitis.
Hypophysitis is defined by inflammation of the pituitary gland and/or infundibulum. It can cause headaches and symptoms due to hypopituitarism. Diagnosis is based on a combination of hormonal and imaging parameters but can, in individuals with an uncertain diagnosis, require a transsphenoidal biopsy. While the risk of immune checkpoint inhibitor-induced hypophysitis is well known, several aetiologies of secondary hypophysitis have been identified and hypophysitis can also occur in relation to pregnancy. Primary hypophysitis is defined by the absence of a secondary cause of hypophysitis. Although each subtype can present with distinct clinical, endocrine and radiological features, extensive overlap makes the aetiological diagnosis difficult. Management of hypophysitis is another challenging task. Whereas some studies have suggested that high-dose glucocorticoids can improve pituitary function and alleviate symptoms, others have found limited benefit. Immunosuppressive agents have also been used as targeted therapy for the underlying condition or in individuals with aggressive hypophysitis, potentially improving both clinical presentation and hormonal function. These therapeutic approaches, used alone or in combination, can be considered in selected patients with primary, pregnancy-related or secondary hypophysitis. The aim of this comprehensive Review is to critically analyse the positive and aetiological diagnostic steps and the management of all subtypes of hypophysitis.
Xanthomatous hypophysitis relapsing and remitting over two decades.
Inflammation of the pituitary gland can be primary (without another underlying cause) or secondary (associated with a systemic inflammatory condition). Primary hypophysitis is very rare, among which xanthomatous hypophysitis as a histological type is extremely unusual. A woman in her late 50s presented with recurrent pituitary lesions over 20 years. Her general practitioner had diagnosed panhypopituitarism in her 30s; a decade later, she had presented to ophthalmology with visual loss and restricted visual fields, and a pituitary lesion was found. This recurred several times requiring multiple resections. Histopathology showed atypical inflammation in keeping with xanthomatous hypophysitis; this responded well to corticosteroid therapy. Xanthomatous hypophysitis is a rare form of steroid-responsive primary pituitary inflammation, to consider in the differential diagnosis of recurring pituitary lesions.
Clinical and radiological insights into secondary hypophysitis: A single-center experience with a focus on tuberculosis.
Secondary hypophysitis (apart from immune checkpoint inhibitor [ICI] induced) is rare and is largely described in case series. We aim to describe the distinctive characteristics of the various etiologies of secondary hypophysitis from a single center. A retrospective record review of 44 patients with secondary hypophysitis (excluding ICI) presenting to our institute between January 2002 and January 2023 was performed. The data of primary hypophysitis managed medically (n = 39) was retrieved from a prior publication and compared with common etiologies of secondary hypophysitis. The most common etiologies were histiocytic disorders - Langerhans cell histiocytosis (LCH) and Erdheim Chester disease (ECD) [n = 23] and tubercular hypophysitis (TH) [n = 10]. LCH/ECD were characterized by multisystem involvement, with arginine vasopressin deficiency (AVP-D) [22/23] being the predominant endocrine presentation. TH patients presented with mass effect (9/10), focal non-enhancing areas within an enhancing sellar/suprasellar mass on magnetic resonance imaging (MRI) (10/10), with evidence of tuberculosis elsewhere in 60%. Though caseating granulomas were universal on histopathology, bacteriological confirmation was negative in all pituitary specimens. When compared to primary hypophysitis, isolated infundibuloneurohypophysitis and AVP-D were more prevalent in LCH/ECD, while the presence of a sellar/suprasellar mass with focal non-enhancing areas was more frequent in TH. Furthermore, recovery of the hormonal axis upon follow-up was more common in primary hypophysitis. Secondary hypophysitis in our cohort was predominantly histiocytic or tubercular in etiology, with LCH/ECD presenting largely with AVP-D and TH presenting with mass effects, focal non-enhancing areas, and paucibacillary disease.
Analysis of a series of 14 clinical cases of neurosurgical treatment of hypophysitis.
Primary hypophysitis is a rare disease that is usually diagnosed retrospectively after surgery for suspected tumors of the sellar region (pituitary adenomas, craniopharyngiomas, etc.). The most common variant of the primary forms is lymphocytic hypophysitis, characterized by the presence of lymphocytes in the inflammatory infiltrate. Granulomatous hypophysitis is the second most common variant of the disease, the cause of which remains unknown. To study the frequency and nature of clinical manifestations, the features of MRI of the brain, as well as the results of neurosurgical treatment of patients with a confirmed histological diagnosis of hypophysitis. A retrospective analysis of the case reports of 14 patients with histologically confirmed diagnosis of lymphocytic (13 cases) and granulomatous (1 case) hypophysitis operated at the Burdenko Neurosurgical Center. In none of the cases before the operation, according to the MRI data, the diagnosis of "hypophysitis" was made. Clinical symptoms were manifested by headaches in 12 patients, decreased acuity and/or visual field impairment in 9 patients, oculomotor impairments in 2 patients. Hypopituitarism was detected in 12 cases, and diabetes insipidus in 8 cases. After surgery, 7 patients had a regression of headache and improved vision, in 5 cases there was no dynamics, in 1 case vision deteriorated. In all 8 patients with diabetes insipidus, it persisted after surgery. There were no new cases of diabetes insipidus. Panhypopituitarism was noted in all patients. Given the difficulty of diagnosing hypophysitis without morphological verification, as well as the rarity of these cases, prospective multicenter studies are needed to study the pathognomonic signs of hypophysitis and improve their neuroimaging methods. Первичный гипофизит — редкое заболевание, обычно диагностируемое ретроспективно после операции по поводу предполагаемых опухолей хиазмально-селлярной области (аденомы гипофиза, краниофарингиомы или др.). Наиболее частым вариантом первичных форм является лимфоцитарный гипофизит, характеризующийся наличием в воспалительном инфильтрате лимфоцитов. Гранулематозный гипофизит — второй по частоте вариант заболевания, причина которого остается неизвестной. Изучить частоту и характер клинических проявлений, особенности МРТ головного мозга, а также результаты нейрохирургического лечения пациентов с подтвержденным гистологическим диагнозом гипофизита. Проведен ретроспективный анализ историй болезни 14 пациентов с гистологически подтвержденным диагнозом лимфоцитарный (13 случаев) и гранулематозный (1 случай) гипофизит, оперированных в НМИЦ нейрохирургии им. акад. Н.Н. Бурденко МЗ РФ. Ни в одном случае до операции по данным МРТ диагноз «гипофизит» поставлен не был. Клиническая симптоматика проявлялась головными болями у 12 пациентов, снижением остроты и/или нарушением полей зрения — у 9, глазодвигательными нарушениями — у 2 пациентов. В 12 случаях был выявлен гипопитуитаризм, в 8 — несахарный диабет. После операции у 7 пациентов отмечался регресс головной боли и улучшение зрения, в 5 случаях динамики не было, в 1 случае зрение ухудшилось. У всех 8 пациентов с несахарным диабетом он сохранился и после операции. Новых случаев несахарного диабета не было. У всех пациентов отмечен пангипопитуитаризм. Учитывая трудность диагностики гипофизитов без морфологической верификации, а также редкость этих образований, необходимы проспективные многоцентровые исследования для изучения патогномоничных признаков гипофизитов и усовершенствования методов их нейровизуализации.
Publicações recentes
Lymphocytic Hypophysitis.
Primary hypophysitis: Classification review.
An update on hypophysitis.
The etiological diagnosis of "Primary" hypophysitis requires prolonged follow-up: A case of Langerhans cell histiocytosis.
📚 EuropePMC32 artigos no totalmostrando 53
Primary hypophysitis: Classification review.
Journal of neuropathology and experimental neurologyAn update on hypophysitis.
Nature reviews. EndocrinologyThe etiological diagnosis of "Primary" hypophysitis requires prolonged follow-up: A case of Langerhans cell histiocytosis.
Annales d'endocrinologieClinical and radiological insights into secondary hypophysitis: A single-center experience with a focus on tuberculosis.
EndocrineAnalysis of a series of 14 clinical cases of neurosurgical treatment of hypophysitis.
Zhurnal voprosy neirokhirurgii imeni N. N. BurdenkoXanthomatous hypophysitis relapsing and remitting over two decades.
Practical neurologyStalking the stalk: Isolated pituitary stalk thickening and predictive factors for proliferative disease.
Neuro-oncology advancesComplete remission after glucocorticoid therapy in a patient with primary hypophysitis.
Endocrinology, diabetes & metabolism case reportsEvaluation and follow-up of patients diagnosed with hypophysitis: a cohort study.
European journal of endocrinologyBilateral Sixth Nerve Palsy: A Rare Presentation of Primary Hypophysitis.
Cureus[Differential diagnosis and tactics of managing a patient with primary hypophysitis on the example of a clinical case].
Problemy endokrinologiiIs There A Connection Between Primary Hypophysitis and Celiac Disease?
Experimental and clinical endocrinology & diabetes : official journal, German Society of Endocrinology [and] German Diabetes Association[Primary hypophysitis: diagnosis and treatment multicenter study].
MedicinaUnveiling the Etiopathogenic Spectrum of Hypophysitis: A Narrative Review.
Journal of personalized medicineA novel approach to hypophysitis: outcomes using non-glucocorticoid immunosuppressive therapy.
European journal of endocrinologyA unique coexistence of a plurihormonal pituitary adenoma with granulomatous hypophysitis.
Indian journal of pathology & microbiologyExecutive summary of the consensus document on hypophysitis of the Neuroendocrinology Area of Knowledge of the Spanish Society of Endocrinology and Nutrition.
Endocrinologia, diabetes y nutricionHypophysitis - A Review of Fourteen Cases.
Neurology IndiaGlucocorticoid therapy as first-line treatment in primary hypophysitis: a systematic review and individual patient data meta-analysis.
Endocrine connectionsRecurrent autoimmune hypophysitis treated with rituximab: a case report.
Journal of medical case reportsXanthomatous Hypophysitis: A Case Report and Comprehensive Literature Review.
Frontiers in endocrinologyDifferences between immunotherapy-induced and primary hypophysitis-a multicenter retrospective study.
PituitaryHypophysitis secondary to pembrolizumab: a case report and review of the literature.
Anti-cancer drugsXanthomatous Hypophysitis Presenting in an Adolescent Girl: A Long-Term Follow-Up of a Rare Case and Review of the Literature.
AACE clinical case reportsTh17 Cells Contribute to the Pathology of Autoimmune Hypophysitis.
Journal of immunology (Baltimore, Md. : 1950)Early Pulse Glucocorticoid Therapy and Improved Hormonal Outcomes in Primary Hypophysitis.
NeuroendocrinologyAnti-pituitary antibodies as a marker of autoimmunity in pituitary glands.
Endocrine journalPrimary and Ipilimumab-induced Hypophysitis: A Single-center Case Series.
Endocrine researchClinical Characteristics of Primary Hypophysitis - A Single-Centre Series of 60 Cases.
Experimental and clinical endocrinology & diabetes : official journal, German Society of Endocrinology [and] German Diabetes AssociationA Remarkable Response of Granulomatous Hypophysitis to Infliximab in a Patient With a Background of Crohn's Disease-A Case Report.
Frontiers in endocrinologyClinical Characteristics, Management, and Treatment Outcomes of Primary Hypophysitis: A Monocentric Cohort.
Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolismeRecurring Primary Xanthomatous Hypophysitis Behaving Like Pituitary Adenoma: Additional Case and Literature Review.
World neurosurgeryHypophysitis induced by immune checkpoint inhibitors: a 10-year assessment.
Expert review of endocrinology & metabolismAutoimmune phenomena involving the pituitary gland in children: New developing data about diagnosis and treatment.
Autoimmunity reviewsXanthomatous hypophysitis: A rare case report with review of literature.
Indian journal of pathology & microbiologyHypophysitis - new insights into diagnosis and treatment.
Endokrynologia PolskaIntracranial Germinoma Masquerading as Secondary Granulomatous Hypophysitis: A Case Report and Review of Literature.
NeuroendocrinologyPrimary hypophysitis: Experience of a Single Tertiary Center.
Experimental and clinical endocrinology & diabetes : official journal, German Society of Endocrinology [and] German Diabetes AssociationDisease heterogeneity in IgG4-related hypophysitis: report of two histopathologically proven cases and review of the literature.
Virchows Archiv : an international journal of pathologyPrimary hypophysitis and other autoimmune disorders of the sellar and suprasellar regions.
Reviews in endocrine & metabolic disordersIdiopathic granulomatous hypophysitis: A report of an uncommon disorder.
Indian journal of pathology & microbiologyMECHANISMS IN ENDOCRINOLOGY: Hypophysitis: diagnosis and treatment.
European journal of endocrinologyClinical, Endocrine and Imaging Characteristics of Patients with Primary Hypophysitis.
Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolismeHuman leucocyte antigens coeliac haplotypes and primary autoimmune hypophysitis in caucasian patients.
Clinical endocrinologyIntratumoral granulomatous reaction in recurrent pituitary adenoma: A unique presentation.
Journal of cancer research and therapeuticsIgG4-related hypophysitis is highly prevalent among cases of histologically confirmed hypophysitis.
Brain pathology (Zurich, Switzerland)Xanthomatous hypophysitis associated with autoimmune disease in an elderly patient: A rare case report.
Surgical neurology internationalA complicated case of primary hypophysitis with bilateral intracavernous carotid artery occlusion.
Hormones (Athens, Greece)A study of primary hypophysitis, i.e., two cases of lymphocytic hypophysitis and one IgG4-related variant: The importance of measuring serum IgG4 levels to allow early diagnosis and prompt treatment for the IgG4-related variant.
Journal of the Formosan Medical Association = Taiwan yi zhiThe management of hypophysitis.
Minerva endocrinologicaDiagnosis of Primary Hypophysitis in Germany.
The Journal of clinical endocrinology and metabolismTreatment of Primary Hypophysitis in Germany.
The Journal of clinical endocrinology and metabolismXanthomatous hypophysitis.
Journal of clinical neuroscience : official journal of the Neurosurgical Society of AustralasiaAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Deficiência da hormona pituitária de origem autoimune.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Deficiência da hormona pituitária de origem autoimune
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Primary hypophysitis: Classification review.
- An update on hypophysitis.
- Xanthomatous hypophysitis relapsing and remitting over two decades.
- Clinical and radiological insights into secondary hypophysitis: A single-center experience with a focus on tuberculosis.
- Analysis of a series of 14 clinical cases of neurosurgical treatment of hypophysitis.
- Lymphocytic Hypophysitis.
- Hypophysitis.
- The etiological diagnosis of "Primary" hypophysitis requires prolonged follow-up: A case of Langerhans cell histiocytosis.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:95506(Orphanet)
- MONDO:0019835(MONDO)
- GARD:19281(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Q4826342(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
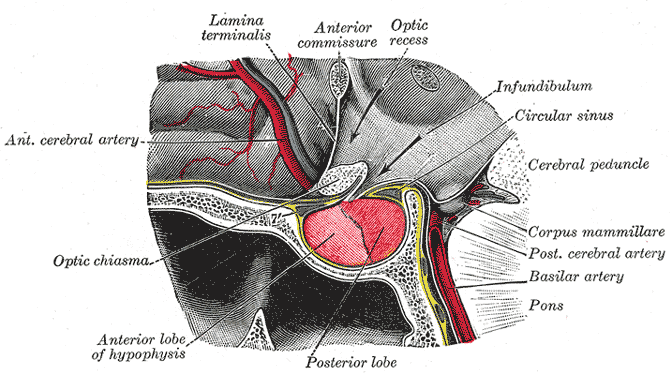
Deficiência da hormona pituitária de origem autoimune
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata