Introdução
O que você precisa saber de cara
Visão geral
A doença monogênica com epilepsia é uma condição genética rara caracterizada pela presença de crises epilépticas (epilepsia) causadas por alterações em um único gene. Isso significa que a epilepsia não é o resultado de múltiplos fatores, mas sim de uma mutação específica em um gene que desempenha um papel crucial no funcionamento do sistema nervoso. Cada caso é único, pois o gene envolvido e o tipo de mutação podem influenciar diretamente os sintomas e a gravidade da doença.[1][3]
Sinais e sintomas
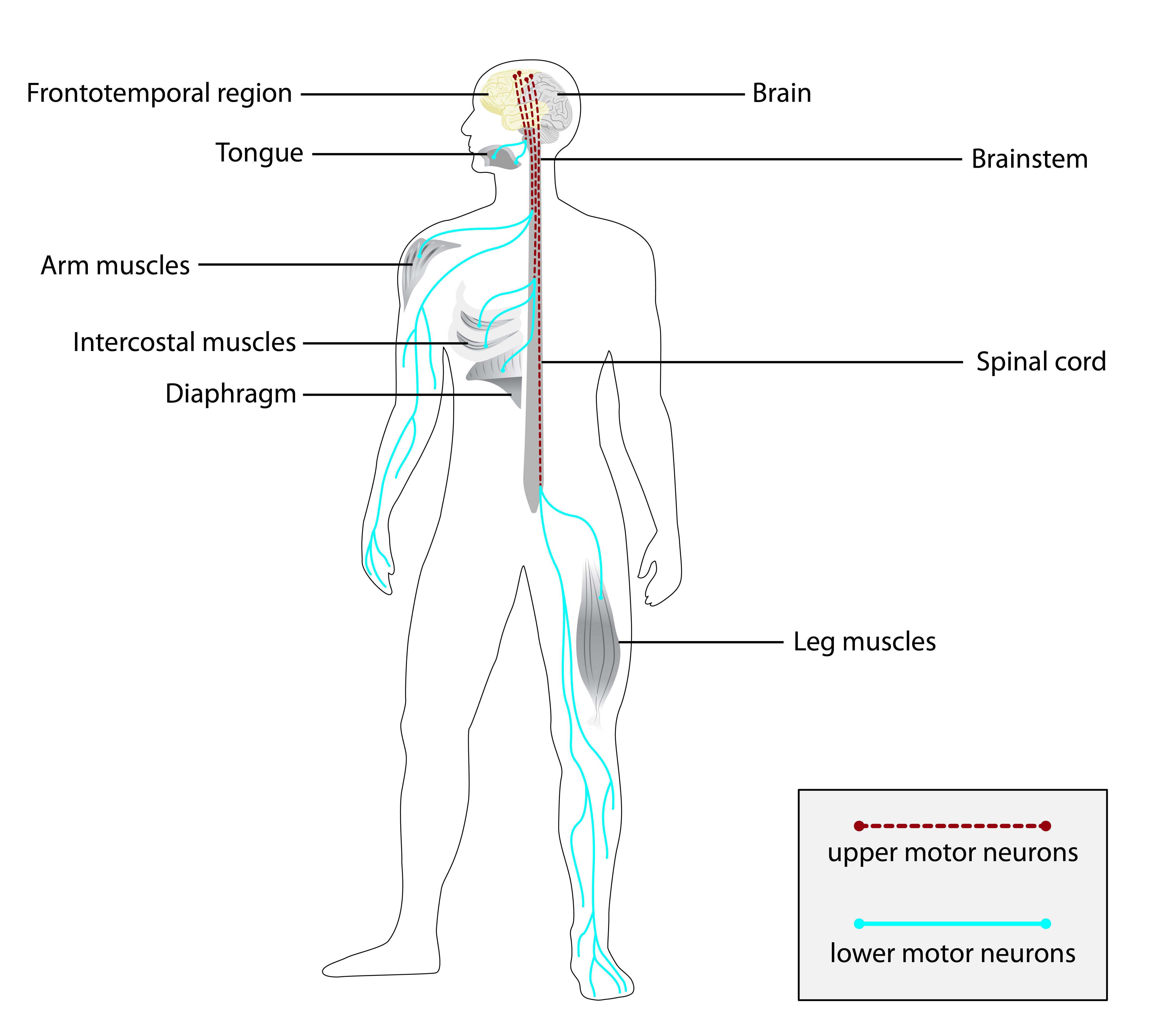
Os sinais e sintomas da doença monogênica com epilepsia são variados e podem afetar diferentes partes do corpo. Além das crises epilépticas, que podem ser detectadas por alterações no eletroencefalograma (EEG), como espículas focais temporais, a condição pode incluir dificuldades para andar (distúrbio da marcha), instabilidade ao ficar em pé (instabilidade postural) e atraso no desenvolvimento da fala (atraso na linguagem expressiva). Outros sintomas possíveis são vômitos episódicos, desconforto respiratório e infecções graves (sepse). Alterações físicas também podem estar presentes, como bossas frontais (proeminência na testa), dorso nasal côncavo (formato do nariz), proptose (olhos saltados), hipermobilidade das articulações dos dedos, displasia ungueal (alterações nas unhas) e edema no dorso dos pés (inchaço). Problemas renais, como duplicação da pelve renal, e malformações ósseas, como sinostose unicoronal direita (fusão precoce dos ossos do crânio), também foram descritos.[1][3]
Causas genéticas
A doença é causada por mutações em um de vários genes. Os genes já identificados incluem: TDP2, TNK2, SCN8A, CPLX1, HACE1, BRAT1, CLN8, DOCK7, CDKL5, PCDH19, SYN1 e GABRA3. Cada um desses genes fornece instruções para a produção de proteínas essenciais para o funcionamento dos neurônios e a transmissão de sinais elétricos no cérebro. Uma alteração em qualquer um deles pode interromper esse processo delicado, levando ao aparecimento da epilepsia.[1][4]
Diagnóstico
O diagnóstico da doença monogênica com epilepsia é baseado na avaliação clínica dos sintomas e, principalmente, na confirmação por meio de testes genéticos. Existem mais de 330 tipos de testes genéticos disponíveis para investigar as alterações nos genes associados a esta condição. Atualmente, mais de 880 variantes genéticas diferentes (mutações) já foram registradas em bancos de dados científicos (ClinVar), o que demonstra a diversidade genética da doença. O diagnóstico genético é fundamental para confirmar a causa da epilepsia e orientar o aconselhamento familiar.[1][4]
Tratamento e manejo
O tratamento da doença monogênica com epilepsia é individualizado e foca no controle das crises epilépticas e no manejo dos sintomas associados. Não há uma cura única, e as abordagens variam conforme o gene envolvido e as necessidades de cada paciente. O acompanhamento deve ser multidisciplinar, envolvendo neurologistas, geneticistas e outros especialistas, para oferecer o melhor suporte possível. É importante lembrar que as informações sobre medicamentos aqui apresentadas são baseadas em estudos e literatura científica, e não substituem a orientação médica personalizada.[1]
Tratamentos citados na literatura
A literatura científica (fonte: PubTator3) menciona diversos fármacos que foram estudados ou associados ao tratamento da epilepsia em contextos genéticos. É fundamental entender que estas são associações mineradas de publicações científicas e não representam uma recomendação de tratamento para todos os casos. Entre as substâncias citadas estão: aminohydroxybutyric-acid, diclofenamide, ethosuximide, felbamate, fosphenytoin, lamotrigine, metharbital, phenacemide, pyridoxal, stiripentol, topiramate, valproic-acid, valpromide e zonisamide. A decisão sobre qual medicamento utilizar deve ser sempre tomada pelo médico, com base no perfil genético e clínico de cada paciente.[1]
Prognóstico e qualidade de vida
O prognóstico para pessoas com doença monogênica com epilepsia é muito variável e depende de fatores como o gene específico afetado, o tipo de mutação, a idade de início dos sintomas e a resposta ao tratamento. Alguns pacientes podem ter crises bem controladas com medicamentos e levar uma vida com boa qualidade, enquanto outros podem apresentar crises mais frequentes e desafios adicionais, como atrasos no desenvolvimento. O acompanhamento médico regular e o suporte multidisciplinar são essenciais para otimizar o desenvolvimento e o bem-estar do paciente e de sua família.[1][3]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Doença monogênica rara caracterizada por epilepsia, distúrbios neurológicos como instabilidade postural e do sistema nervoso, e achados como duplicação renal e displasia ungueal. Associada a mutações em genes como TDP2, TNK2 e SCN8A.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A doença monogênica com epilepsia é uma condição genética rara caracterizada pela presença de crises epilépticas (epilepsia) causadas por alterações em um único gene. Isso significa que a epilepsia não é o resultado de múltiplos fatores, mas sim de uma mutação específica em um gene que desempenha um papel crucial no funcionamento do sistema nervoso. Cada caso é único, pois o gene envolvido e o tipo de mutação podem influenciar diretamente os sintomas e a gravidade da doença.[1][3]
Sinais e sintomas
Os sinais e sintomas da doença monogênica com epilepsia são variados e podem afetar diferentes partes do corpo. Além das crises epilépticas, que podem ser detectadas por alterações no eletroencefalograma (EEG), como espículas focais temporais, a condição pode incluir dificuldades para andar (distúrbio da marcha), instabilidade ao ficar em pé (instabilidade postural) e atraso no desenvolvimento da fala (atraso na linguagem expressiva). Outros sintomas possíveis são vômitos episódicos, desconforto respiratório e infecções graves (sepse). Alterações físicas também podem estar presentes, como bossas frontais (proeminência na testa), dorso nasal côncavo (formato do nariz), proptose (olhos saltados), hipermobilidade das articulações dos dedos, displasia ungueal (alterações nas unhas) e edema no dorso dos pés (inchaço). Problemas renais, como duplicação da pelve renal, e malformações ósseas, como sinostose unicoronal direita (fusão precoce dos ossos do crânio), também foram descritos.[1][3]
Causas genéticas
A doença é causada por mutações em um de vários genes. Os genes já identificados incluem: TDP2, TNK2, SCN8A, CPLX1, HACE1, BRAT1, CLN8, DOCK7, CDKL5, PCDH19, SYN1 e GABRA3. Cada um desses genes fornece instruções para a produção de proteínas essenciais para o funcionamento dos neurônios e a transmissão de sinais elétricos no cérebro. Uma alteração em qualquer um deles pode interromper esse processo delicado, levando ao aparecimento da epilepsia.[1][4]
Diagnóstico
O diagnóstico da doença monogênica com epilepsia é baseado na avaliação clínica dos sintomas e, principalmente, na confirmação por meio de testes genéticos. Existem mais de 330 tipos de testes genéticos disponíveis para investigar as alterações nos genes associados a esta condição. Atualmente, mais de 880 variantes genéticas diferentes (mutações) já foram registradas em bancos de dados científicos (ClinVar), o que demonstra a diversidade genética da doença. O diagnóstico genético é fundamental para confirmar a causa da epilepsia e orientar o aconselhamento familiar.[1][4]
Tratamento e manejo
O tratamento da doença monogênica com epilepsia é individualizado e foca no controle das crises epilépticas e no manejo dos sintomas associados. Não há uma cura única, e as abordagens variam conforme o gene envolvido e as necessidades de cada paciente. O acompanhamento deve ser multidisciplinar, envolvendo neurologistas, geneticistas e outros especialistas, para oferecer o melhor suporte possível. É importante lembrar que as informações sobre medicamentos aqui apresentadas são baseadas em estudos e literatura científica, e não substituem a orientação médica personalizada.[1]
Tratamentos citados na literatura
A literatura científica (fonte: PubTator3) menciona diversos fármacos que foram estudados ou associados ao tratamento da epilepsia em contextos genéticos. É fundamental entender que estas são associações mineradas de publicações científicas e não representam uma recomendação de tratamento para todos os casos. Entre as substâncias citadas estão: aminohydroxybutyric-acid, diclofenamide, ethosuximide, felbamate, fosphenytoin, lamotrigine, metharbital, phenacemide, pyridoxal, stiripentol, topiramate, valproic-acid, valpromide e zonisamide. A decisão sobre qual medicamento utilizar deve ser sempre tomada pelo médico, com base no perfil genético e clínico de cada paciente.[1]
Prognóstico e qualidade de vida
O prognóstico para pessoas com doença monogênica com epilepsia é muito variável e depende de fatores como o gene específico afetado, o tipo de mutação, a idade de início dos sintomas e a resposta ao tratamento. Alguns pacientes podem ter crises bem controladas com medicamentos e levar uma vida com boa qualidade, enquanto outros podem apresentar crises mais frequentes e desafios adicionais, como atrasos no desenvolvimento. O acompanhamento médico regular e o suporte multidisciplinar são essenciais para otimizar o desenvolvimento e o bem-estar do paciente e de sua família.[1][3]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 182 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 503 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
24 genes identificados com associação a esta condição.
DNA repair enzyme that can remove a variety of covalent adducts from DNA through hydrolysis of a 5'-phosphodiester bond, giving rise to DNA with a free 5' phosphate. Catalyzes the hydrolysis of dead-end complexes between DNA and the topoisomerase 2 (TOP2) active site tyrosine residue. The 5'-tyrosyl DNA phosphodiesterase activity can enable the repair of TOP2-induced DNA double-strand breaks/DSBs without the need for nuclease activity, creating a 'clean' DSB with 5'-phosphate termini that are re
NucleusNucleus, PML bodyNucleus, nucleolusCytoplasm
Spinocerebellar ataxia, autosomal recessive, 23
A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCAR23 patients manifest epilepsy, intellectual disability, and gait ataxia.
Non-receptor tyrosine-protein and serine/threonine-protein kinase that is implicated in cell spreading and migration, cell survival, cell growth and proliferation. Transduces extracellular signals to cytosolic and nuclear effectors. Phosphorylates AKT1, AR, MCF2, WASL and WWOX. Implicated in trafficking and clathrin-mediated endocytosis through binding to epidermal growth factor receptor (EGFR) and clathrin. Binds to both poly- and mono-ubiquitin and regulates ligand-induced degradation of EGFR,
Cell membraneNucleusEndosomeCell junction, adherens junctionCytoplasmic vesicle membraneCytoplasmic vesicle, clathrin-coated vesicleMembrane, clathrin-coated pitCytoplasm, perinuclear regionCytoplasm, cytosol
Pore-forming subunit of a voltage-gated sodium channel complex assuming opened or closed conformations in response to the voltage difference across membranes and through which sodium ions selectively pass along their electrochemical gradient (PubMed:24874546, PubMed:25239001, PubMed:25725044, PubMed:26900580, PubMed:29726066, PubMed:33245860, PubMed:36696443, PubMed:36823201). Contributes to neuronal excitability by regulating action potential threshold and propagation (PubMed:24874546, PubMed:2
Cell membraneCell projection, axonCytoplasmic vesicleCell projection, podosome
Cognitive impairment with or without cerebellar ataxia
A disorder characterized by markedly delayed cognitive and motor development, attention deficit disorder, and cerebellar ataxia. Features include bilateral esophoria, strabismatic amblyopia, unsustained gaze evoked nystagmus on horizontal gaze, ataxic gait, dysmetria in the upper limbs and dysarthria, with normal strength, tone, and reflexes.
Positively regulates a late step in exocytosis of various cytoplasmic vesicles, such as synaptic vesicles and other secretory vesicles (PubMed:21785414). Organizes the SNAREs into a cross-linked zigzag topology that, when interposed between the vesicle and plasma membranes, is incompatible with fusion, thereby preventing SNAREs from releasing neurotransmitters until an action potential arrives at the synapse (PubMed:21785414). Also involved in glucose-induced secretion of insulin by pancreatic b
Cytoplasm, cytosolPerikaryonPresynapse
Developmental and epileptic encephalopathy 63
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE63 is an autosomal recessive disease with onset in infancy.
E3 ubiquitin-protein ligase involved in Golgi membrane fusion and regulation of small GTPases (PubMed:15254018, PubMed:21988917, PubMed:22036506, PubMed:37537642, PubMed:38332367). Acts as a regulator of Golgi membrane dynamics during the cell cycle: recruited to Golgi membrane by Rab proteins and regulates postmitotic Golgi membrane fusion (PubMed:21988917). Acts by mediating ubiquitination during mitotic Golgi disassembly, ubiquitination serving as a signal for Golgi reassembly later, after ce
Golgi apparatus, Golgi stack membraneCytoplasmEndoplasmic reticulum
Component of a multiprotein complex required for the assembly of the RNA endonuclease module of the integrator complex (PubMed:39032489, PubMed:39032490). Associates with INTS9 and INTS11 in the cytoplasm and blocks the active site of INTS11 to inhibit the endonuclease activity of INTS11 before formation of the full integrator complex (PubMed:39032489, PubMed:39032490). Following dissociation of WDR73 of the complex, BRAT1 facilitates the nuclear import of the INTS9-INTS11 heterodimer (PubMed:39
NucleusCytoplasm
Rigidity and multifocal seizure syndrome, lethal neonatal
A lethal, neonatal, neurologic disorder characterized by episodic jerking that is apparent in utero, lack of psychomotor development, axial and limb rigidity, frequent multifocal seizures, and dysautonomia. At birth, affected individuals have small heads, overlapping cranial sutures, small or absent fontanels, and depressed frontal bones. Infants show poorly responsive focal jerks of the tongue, face and arms in a nearly continuous sequence throughout life.
Could play a role in cell proliferation during neuronal differentiation and in protection against cell death
Endoplasmic reticulum membraneEndoplasmic reticulum-Golgi intermediate compartment membraneEndoplasmic reticulum
Ceroid lipofuscinosis, neuronal, 8
A form of neuronal ceroid lipofuscinosis with onset in childhood. Neuronal ceroid lipofuscinoses are progressive neurodegenerative, lysosomal storage diseases characterized by intracellular accumulation of autofluorescent liposomal material, and clinically by seizures, dementia, visual loss, and/or cerebral atrophy. The lipopigment patterns observed most often in neuronal ceroid lipofuscinosis type 8 comprise mixed combinations of granular, curvilinear, and fingerprint profiles.
Functions as a guanine nucleotide exchange factor (GEF), which activates Rac1 and Rac3 Rho small GTPases by exchanging bound GDP for free GTP. Does not have a GEF activity for CDC42. Required for STMN1 'Ser-15' phosphorylation during axon formation and consequently for neuronal polarization (PubMed:16982419). As part of the DISP complex, may regulate the association of septins with actin and thereby regulate the actin cytoskeleton (PubMed:29467281). Has a role in pigmentation (By similarity). In
Cell projection, axon
Developmental and epileptic encephalopathy 23
A severe disease characterized by early-onset intractable epilepsy, dysmorphic features, intellectual disability, and cortical blindness. Brain imaging shows an abnormally marked pontobulbar sulcus with mild pontine hypoplasia, white matter abnormalities, and atrophy in the occipital lobe.
Mediates phosphorylation of MECP2 (PubMed:15917271, PubMed:16935860). May regulate ciliogenesis (PubMed:29420175)
NucleusCytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, microtubule organizing center, centrosome
Calcium-dependent cell-adhesion protein
Cell membrane
Developmental and epileptic encephalopathy 9
A condition characterized by seizure with onset in infancy or early childhood, cognitive impairment, and delayed development of variable severity in some patients. Additional features include autistic signs and psychosis. The disorder is sex-limited, with the phenotype being restricted to females.
Neuronal phosphoprotein that coats synaptic vesicles, and binds to the cytoskeleton. Acts as a regulator of synaptic vesicles trafficking, involved in the control of neurotransmitter release at the pre-synaptic terminal (PubMed:21441247, PubMed:23406870). Also involved in the regulation of axon outgrowth and synaptogenesis (By similarity). The complex formed with NOS1 and CAPON proteins is necessary for specific nitric-oxid functions at a presynaptic level (By similarity)
SynapseGolgi apparatusPresynapseCytoplasmic vesicle, secretory vesicle, synaptic vesicle
Epilepsy, X-linked 1, with variable learning disabilities and behavior disorders
A neurologic disorder characterized by variable combinations of epilepsy, learning difficulties, macrocephaly, and aggressive behavior.
Alpha subunit of the heteropentameric ligand-gated chloride channel gated by gamma-aminobutyric acid (GABA), a major inhibitory neurotransmitter in the brain (PubMed:16412217, PubMed:29053855). GABA-gated chloride channels, also named GABA(A) receptors (GABAAR), consist of five subunits arranged around a central pore and contain GABA active binding site(s) located at the alpha and beta subunit interface(s) (By similarity). When activated by GABA, GABAARs selectively allow the flow of chloride an
Postsynaptic cell membraneCell membrane
Epilepsy, X-linked 2, with or without impaired intellectual development and dysmorphic features
A neurologic disorder characterized by variable combinations of epileptic seizure, and a varying degree of intellectual disability and developmental delay. Some patients have dysmorphic facial features or mild skeletal anomalies. In general, males are more severely affected than females, although there is evidence for incomplete penetrance in both sexes.
E3 ubiquitin-protein ligase that regulates cell proliferation (PubMed:18794910, PubMed:23378536, PubMed:30595371). Involved in apoptosis regulation (PubMed:23378536, PubMed:30595371). Mediates ER stress-induced activation of JNK signaling pathway and apoptosis by promoting ERN1 activation and splicing of XBP1 mRNA (PubMed:23378536, PubMed:30595371). Also involved in protein trafficking and localization (PubMed:24387786)
Endoplasmic reticulum membraneLate endosome membraneLysosome membraneNucleus inner membrane
Developmental and epileptic encephalopathy 73
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE73 is an autosomal dominant form with onset at birth.
Chromosomal protein that binds to methylated DNA. It can bind specifically to a single methyl-CpG pair. It is not influenced by sequences flanking the methyl-CpGs. Mediates transcriptional repression through interaction with histone deacetylase and the corepressor SIN3A. Binds both 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC)-containing DNA, with a preference for 5-methylcytosine (5mC)
Nucleus
Angelman syndrome
A neurodevelopmental disorder characterized by severe motor and intellectual retardation, ataxia, frequent jerky limb movements and flapping of the arms and hands, hypotonia, seizures, absence of speech, frequent smiling and episodes of paroxysmal laughter, open-mouthed expression revealing the tongue.
Deubiquitinating enzyme that may play a role in the ubiquitin-dependent regulation of protein synthesis, downstream of mTORC1 (PubMed:21267069, PubMed:27864334). May associate with the protein synthesis initiation complex and modify its ubiquitination to repress translation (PubMed:27864334). May also repress DNA synthesis and modify different cellular targets thereby regulating cell growth and proliferation (PubMed:27864334). May also play a role in proteasome assembly and function (PubMed:2834
Intellectual developmental disorder with dysmorphic facies, seizures, and distal limb anomalies
An autosomal recessive severe multisystem disorder characterized by poor overall growth, developmental delay, early-onset seizures, intellectual disability, and dysmorphic features. Additional features include microcephaly, absent speech, hypotonia, feeding difficulties, structural brain abnormalities, congenital malformations including congenital heart disease, and musculoskeletal features.
Acts as a guanine nucleotide exchange factor (GEF) for CDC42. Promotes formation of GPHN clusters (By similarity)
CytoplasmPostsynaptic density
Developmental and epileptic encephalopathy 8
A disorder characterized by hyperekplexia and early infantile epileptic encephalopathy. Neurologic features include exaggerated startle response, seizures, impaired psychomotor development, and intellectual disability. Seizures can be provoked by tactile stimulation or extreme emotion.
Pseudokinase which, in complex with CAB39/MO25 (CAB39/MO25alpha or CAB39L/MO25beta), binds to and activates STK11/LKB1. Adopts a closed conformation typical of active protein kinases and binds STK11/LKB1 as a pseudosubstrate, promoting conformational change of STK11/LKB1 in an active conformation
NucleusCytoplasm
May act as a GTPase-activating protein for Rab family protein(s) (PubMed:20727515, PubMed:20797691). Involved in neuronal projections development, probably through a negative modulation of ARF6 function (PubMed:20727515). Involved in the regulation of synaptic vesicle trafficking (PubMed:31257402)
Cell membraneCytoplasmCytoplasmic vesicle membranePresynapse
Familial infantile myoclonic epilepsy
A subtype of idiopathic epilepsy starting in early infancy and manifesting as myoclonic seizures, febrile convulsions, and tonic-clonic seizures.
Sequence-specific RNA-binding protein that acts as a post-transcriptional repressor by binding the 3'-UTR of mRNA targets. Binds to an RNA consensus sequence, the Pumilio Response Element (PRE), 5'-UGUANAUA-3', that is related to the Nanos Response Element (NRE) (PubMed:18328718, PubMed:21397187, PubMed:21572425, PubMed:21653694). Mediates post-transcriptional repression of transcripts via different mechanisms: acts via direct recruitment of the CCR4-POP2-NOT deadenylase leading to translational
CytoplasmCytoplasm, P-bodyCytoplasmic granule
Neurodevelopmental disorder with motor abnormalities, seizures, and facial dysmorphism
An autosomal dominant disorder characterized by global developmental delay, impaired intellectual development, early-onset seizures, poor overall growth, delayed walking, hypotonia and/or ataxia, and facial dysmorphism. Some patients have hypoplasia of the corpus callosum and cerebral atrophy.
Transcriptional inhibitor that binds to DNA sequence 5'-CACCT-3' in different promoters (PubMed:16061479, PubMed:20516212). Represses transcription of E-cadherin (PubMed:16061479). Represses expression of MEOX2 (PubMed:20516212)
NucleusChromosome
Mowat-Wilson syndrome
A complex developmental disorder characterized by intellectual disability, delayed motor development, epilepsy, microcephaly and a wide spectrum of clinically heterogeneous features suggestive of neurocristopathies at the cephalic, cardiac, and vagal levels. Affected patients show an easily recognizable facial appearance with deep set eyes and hypertelorism, medially divergent, broad eyebrows, prominent columella, pointed chin and uplifted, notched ear lobes. Some patients manifest Hirschsprung disease.
Multifunctional protein which functions as a renin, prorenin cellular receptor and is involved in the assembly of the lysosomal proton-transporting V-type ATPase (V-ATPase) and the acidification of the endo-lysosomal system (PubMed:12045255, PubMed:29127204, PubMed:30374053, PubMed:32276428). May mediate renin-dependent cellular responses by activating ERK1 and ERK2 (PubMed:12045255). By increasing the catalytic efficiency of renin in AGT/angiotensinogen conversion to angiotensin I, may also pla
Endoplasmic reticulum membraneLysosome membraneCytoplasmic vesicle, autophagosome membraneCell projection, dendritic spine membraneCell projection, axonEndosome membraneCytoplasmic vesicle, clathrin-coated vesicle membraneCytoplasmic vesicle, secretory vesicle, synaptic vesicle membrane
Intellectual developmental disorder, X-linked, syndromic, Hedera type
A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSH patients manifest mild to moderate intellectual disability associated with epilepsy, delays in motor milestones and speech acquisition in infancy.
Inhibits PIK3C3 activity; under basal conditions negatively regulates PI3K complex II (PI3KC3-C2) function in autophagy. Negatively regulates endosome maturation and degradative endocytic trafficking and impairs autophagosome maturation process. Can sequester UVRAG from association with a class C Vps complex (possibly the HOPS complex) and negatively regulates Rab7 activation (PubMed:20974968, PubMed:21062745) Involved in regulation of pathogen-specific host defense of activated macrophages. Fol
Late endosomeLysosomeEarly endosome
Spinocerebellar ataxia, autosomal recessive, 15
A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCAR15 patients manifest cerebellar ataxia in early childhood and delayed motor development with delayed walking. Additional features include dysarthria, upper limb involvement, abnormal eye movements, and hyporeflexia.
Putative oxidoreductase. Acts as a tumor suppressor and plays a role in apoptosis. Required for normal bone development (By similarity). May function synergistically with p53/TP53 to control genotoxic stress-induced cell death. Plays a role in TGFB1 signaling and TGFB1-mediated cell death. May also play a role in tumor necrosis factor (TNF)-mediated cell death. Inhibits Wnt signaling, probably by sequestering DVL2 in the cytoplasm
CytoplasmNucleusMitochondrionGolgi apparatusLysosome
Functions as a transcriptional repressor. Seems to function as a transcriptional corepressor in neuronal cells through recruitment of HDAC3 and HDAC4. Contributes to tumor progression, and tumor cell proliferation and survival. This may be mediated at least in part through repressing transcriptional activity of GADD45GIP1. Required for recruiting the proteasome from the nucleus to the cytoplasm and dendritic spines
NucleusCytoplasm
Neurodevelopmental disorder with epilepsy, cataracts, feeding difficulties, and delayed brain myelination
A neurodevelopmental disorder characterized by microcephaly, profound developmental delay, intellectual disability, cataracts, severe epilepsy including infantile spasms, irritability, failure to thrive, and stereotypic hand movements. Brain imaging reveals delayed myelination and cerebral atrophy.
Variantes genéticas (ClinVar)
888 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
51 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Doença monogênica com epilepsia
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
Characterizing SCN1A -Related Disorders Using Real-World Data Across 681 Patient-Years.
SCN1A -related disorders are the single most common monogenic cause of epilepsy and represent a major focus of precision medicine efforts. In conjunction with existing prospective studies, the analysis of real-world data obtained during routine clinical care can expand upon the scale and duration of available data and contribute to the development of meaningful outcomes for clinical trials. Here, we leveraged real-world data to delineate the longitudinal disease history of 100 individuals with SCN1A -related disorders using a systematic approach. We mapped a total of 671 unique clinical terms to a standardized framework in monthly increments across 681 patient-years, including 75 terms related to seizure types. Within this cohort, 89 individuals had presumed loss-of-function variants in SCN1A based on variant type and clinical diagnosis, including those with Dravet syndrome ( N = 79) and genetic epilepsy with febrile seizures plus ( N = 10). Ten individuals had a non-Dravet developmental and epileptic encephalopathy caused by gain-of-function variants in SCN1A . By annotating seizure type and frequency in monthly time-bins, we assessed seizure burden. A median of 17 changes in seizure frequency and ten terms referring to seizure type were identified per participant. Myoclonic seizures occurred with high frequency (median >5 daily), whereas hemiclonic, focal impaired consciousness, and bilateral tonic-clonic seizures occurred more rarely (median monthly). Retrospective analysis of developmental histories showed a range of cognitive abilities. Neurodevelopmental differences were observed in 83% (83/100) of individuals, of whom 83% (69/83) demonstrated delayed language skills. Motor coordination impairments, including gait disturbance, ataxia, hypotonia, and imbalance were annotated in 69% (69/100) of participants. EEG findings varied with age; most were reported as normal before nine months of age, after which the prevalence of abnormal interictal findings increased. Individuals with different clinical syndromes had unique medication landscapes, with 554 prescriptions of 37 unique therapies. Changes in treatment coincided with the diagnosis of an SCN1A -related disorder, with an increase in cannabidiol, clobazam, and fenfluramine and reduction in sodium channel-blocker use following genetic diagnosis. In summary, we reconstructed the longitudinal disease history of SCN1A -related disorders from electronic medical records using a standardized framework for the analysis of real-world clinical data. We refine existing natural history data of SCN1A -related disorders by providing a granular landscape of seizures, comorbidities, and treatment approaches over time.
Mitochondrial Dysfunction in Monogenic Developmental and Epileptic Encephalopathies.
Before diagnostic whole exome sequencing, monogenic/chromosomal developmental and epileptic encephalopathies (DEEs) were frequently misdiagnosed as mitochondrial disorders (MDs) with epilepsy, due to overlapping clinical and biochemical features. Assessing muscle functional assays in patients with a genetic diagnosis and epilepsy offers a unique opportunity to explore mitochondrial dysfunction in monogenic/chromosomal DEEs, in comparison to the mitochondrial dysfunction observed in genetically confirmed MDs. In this retrospective cohort study, clinical and biochemical data were extracted from patients suspected of MD with epilepsy who underwent muscle/fibroblast biopsy (2005-2015). Patients were classified into four groups based on the final diagnosis. Mitochondrial Disease Criteria scores were assigned. Statistical analyses were conducted using Fisher's exact, analysis of variance, and Kruskal-Wallis tests. Of 27 included participants, eleven (40.7%) had DEEs, four (14.8%) had genetically confirmed MDs, eight (29.6%) were suspected MD cases without genetic confirmation, and four (14.8%) had nonmitochondrial metabolic diseases. Mitochondrial dysfunction was common across all groups; 85% of participants met probable/definite Mitochondrial Disease Criteria, over 70% had elevated plasma lactate (>2.5 mmol/L), and 92% exhibited impaired adenosine triphosphate production. Surprisingly, moderate to severe complex dysfunction was observed in all groups except genetically confirmed MDs. Our findings indicate that mitochondrial dysfunction is prevalent in nonmitochondrial DEEs. Patients previously diagnosed with an MD based only on muscle/fibroblast biopsy may benefit from whole exome sequencing to identify genetic variants, for which targeted therapy may be available. Future research should explore whether treatment or prognosis of nonmitochondrial DEEs should be tailored to improve mitochondrial function.
Research progress in SYNGAP1-related neurodevelopmental disorders: from pathogenesis to therapeutic strategies.
SYNGAP1-related neurodevelopmental disorder (SRD) is a monogenic inherited brain disorder caused by heterozygous loss-of-function mutations in the SYNGAP1 gene. The clinical presentation is complex, with core features including global developmental delay/intellectual disability, epilepsy, autism spectrum disorder, and various behavioral abnormalities. The SynGAP protein, encoded by the SYNGAP1 gene, is a key regulatory protein in the postsynaptic density of excitatory neurons. Through its GTPase-activating protein activity and structural scaffolding functions, it plays a central role in regulating the Ras/Rap signaling pathways, AMPA receptor trafficking, and maintaining the excitatory/inhibitory balance of neural networks. Haploinsufficiency of SynGAP leads to synaptic plasticity disruption and neural circuit imbalance, thereby triggering a series of neurophysiological and behavioral phenotypes. This article systematically reviews the molecular pathogenesis of SRDs, summarizes advances in treatment from conventional anti-seizure medications to emerging precision therapeutic strategies such as gene supplementation, antisense oligonucleotide-mediated splicing modulation, and translation-activating RNAs, and discusses current research challenges and future directions. Key concepts central to understanding SRDs include the critical developmental periods during which SynGAP exerts its primary influence on synaptic maturation, and cell-type specificity, referring to the differential expression and function of SynGAP in distinct neuronal populations (e.g., excitatory pyramidal neurons vs. parvalbumin-positive interneurons), which underlies circuit-level dysfunction. The aim is to provide a comprehensive perspective for an in-depth understanding of the disease and to support the development of effective therapies.
International Clinical Evidence-based Guideline for Kleefstra Syndrome.
Kleefstra syndrome (KLEFS1) is a rare monogenic neurodevelopmental disorder (mNDD) with multisystem involvement, caused by disruption of EHMT1 function, resulting in significant burden on affected individuals and their families. The current shortage of and globally scattered syndrome-specific knowledge has led to significant disparities in the access to and provision of evidence-based and individual-centered expert care. To address the challenges and improve outcomes for individuals with KLEFS1, an international KLEFS1 guideline consortium was formed consisting of 43 participants, both clinical experts and patient-representatives, from 15 different countries. The primary goal of the consortium was to develop a comprehensive and high-quality guideline for KLEFS1, aiming to enhance patient care, establish a uniform minimum international standard of care, and support decision-making. The current clinical guideline is evidence-based and includes 66 tailored recommendations to improve KLEFS1 care. The comprehensive methodological approach ensures broad consensus and supports effective implementation. Furthermore, this guideline serves as a valuable methodological model for guideline development in the context of rare disorders.
Seizure outcomes after VNS therapy in children with drug-resistant epilepsy due to monogenic etiologies versus malformations of cortical development.
Vagus nerve stimulation (VNS) is an established adjunctive therapy for drug-resistant epilepsy (DRE). However, evidence regarding its efficacy in children with structural and nonstructural etiologies of epilepsy remains limited. Herein, the authors aimed to explore the effectiveness of VNS in patients with either etiology. In this retrospective single-center study, authors evaluated children (ages 2-18 years) with DRE due to nonlesional monogenic epilepsies (MEs) or malformations of cortical development (MCDs) who had undergone VNS device implantation between 2008 and 2024 and had ≥ 6 months of follow-up. Patients with tuberous sclerosis complex were excluded. Clinical, genetic, neuroimaging, VNS programming, and outcome data were extracted from medical records. The primary outcome was the responder rate, defined as > 50% seizure reduction from baseline at 6 months, 12 months, and the last follow-up, in the two groups. Of 336 VNS device recipients, 64 children with ME (n = 44) or MCD (n = 20) met the study inclusion criteria. The median follow-up was 3.5-4.0 years. The responder rate in the ME versus MCD group at 6 months, 1 year, and the last follow-up was 38.6% versus 42.1% (p = 0.77), 45.7% versus 44.4% (p = 0.46), and 47.6% versus 63.2% (p = 0.64), respectively. The SCN1A-related Dravet syndrome (SCN1A-DS) subgroup (n = 12) had a responder rate (50.0%) comparable to that of the non-SCN1A-DS ME group (43.7%) at the last follow-up. The frequency of status epilepticus decreased significantly in both groups (p = 0.03). VNS was well tolerated, with mild to moderate side effects reported in < 5% patients. No clinical variable, including age at seizure onset, epilepsy duration, or age at VNS device implantation predicted seizure outcomes. VNS therapy was noted to have a similar responder rate in children with DRE due to MEs or MCDs. Both groups experienced meaningful benefits, with a small proportion of patients experiencing mild side effects.
Publicações recentes
Molecular Profiling of Polish Pediatric Patients with Epilepsy: A Single-Center Diagnostic Experience Using Next-Generation Sequencing.
Seizure outcomes after VNS therapy in children with drug-resistant epilepsy due to monogenic etiologies versus malformations of cortical development.
Altered Auditory Maturation in Fragile X Syndrome and Its Involvement in Audiogenic Seizure Susceptibility.
Genetic Variants and Disease Mechanisms: Lessons from Monogenic Childhood Epilepsies.
Clinical and genetic landscape of epilepsies with absence seizures and single-gene etiology.
📚 EuropePMCmostrando 198
Characterizing SCN1A -Related Disorders Using Real-World Data Across 681 Patient-Years.
medRxiv : the preprint server for health sciencesMitochondrial Dysfunction in Monogenic Developmental and Epileptic Encephalopathies.
Pediatric neurologyResearch progress in SYNGAP1-related neurodevelopmental disorders: from pathogenesis to therapeutic strategies.
Frontiers in neurologyMolecular Profiling of Polish Pediatric Patients with Epilepsy: A Single-Center Diagnostic Experience Using Next-Generation Sequencing.
GenesNeurocardiogenetics: Exploring the association of rare RYR2 variants with neuropsychiatric disorders in general and disease populations.
Journal of neurogeneticsGenetic Risk Factors for Epilepsy: From Familial Studies to Gene Discoveries and Polygenic Risk Scores, Strides Toward Unlocking an Age-Old Question in Epilepsy.
Epilepsy currentsAAV-mediated gene therapy in a SLC13A5 citrate transporter disorder model rescues epileptic and metabolic phenotypes.
The Journal of clinical investigationEndoplasmic reticulum protein retention and disturbed proteostasis is a common pathology for a subset of autism: evidence from mutations in GABAA receptors and GABA transporter 1.
Frontiers in molecular neuroscienceInternational Clinical Evidence-based Guideline for Kleefstra Syndrome.
Genetics in medicine : official journal of the American College of Medical GeneticsSeizure outcomes after VNS therapy in children with drug-resistant epilepsy due to monogenic etiologies versus malformations of cortical development.
Journal of neurosurgery. PediatricsAltered Auditory Maturation in Fragile X Syndrome and Its Involvement in Audiogenic Seizure Susceptibility.
Autism research : official journal of the International Society for Autism ResearchAtypical GNAO1 variants in severe childhood speech disorders: clinical, genetic, and molecular insights.
Molecular autismRevealing Monogenic Diabetes: Clinical and Genetic Features of Pediatric MODY Cases in Türkiye: Single Center Experience.
Pediatric diabetesPrecision medicine for sodium channelopathy-related autism and epilepsy.
Trends in molecular medicineGenetic Variants and Disease Mechanisms: Lessons from Monogenic Childhood Epilepsies.
NeuropediatricsClinical and genetic landscape of epilepsies with absence seizures and single-gene etiology.
EpilepsiaGRIN2A null variants confer a high risk for early-onset schizophrenia and other mental disorders and potentially enable precision therapy.
Molecular psychiatryThe LMSz method - an automatable scalable approach to constructing gene-specific growth charts in rare disorders.
European journal of human genetics : EJHGChildren With Pancreatic Hypoplasia Experience Poor Weight Gain and Labile Diabetes but Low Incidence of DKA.
The Journal of clinical endocrinology and metabolismAtenolol rescues premature mortality in genetic mouse models of sudden unexpected death in epilepsy.
EpilepsiaWhole Exome Sequencing Based Diagnostics in Complex Childhood Epilepsy Syndromes-A Cohort Study on Clinical Utility.
Clinical geneticsEpilepsy-Associated SCN2A-L1342P Mutation Drives Network Hyperexcitability and Widespread Transcriptomic Changes in Human Cortical Organoids.
bioRxiv : the preprint server for biologyNeonatal diabetes mellitus is a significant feature of COXPD-24 caused by recessive NARS2 variants.
Diabetic medicine : a journal of the British Diabetic AssociationCSF IL-6 in children with neuroinflammatory conditions.
European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology SocietyELFN1 deficiency: The mechanistic basis and phenotypic spectrum of a neurodevelopmental disorder with epilepsy.
Genetics in medicine : official journal of the American College of Medical GeneticsEmerging Insights into the Pathogenic Mechanisms of Dravet Syndrome.
Neurochemical researchIncreased Activity-Dependent Bulk Endocytosis in Huntington's Disease Results From Huntingtin Haploinsufficiency.
Journal of neurochemistryGenome-Wide Insights and Polygenic Risk Scores in Common Epilepsies: A Narrative Review.
American journal of medical genetics. Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric GeneticsSCN9A should not be considered an epilepsy gene; Refuting a gene-disease association.
EpilepsiaBridging the gap: Canine in vitro models of the central nervous system as tools to study pathogenetic mechanisms in neurodegenerative disease and neuroinfectiology.
Veterinary pathologyPrecision therapies for genetic epilepsies in 2025: Promises and pitfalls.
Epilepsia openConvergent depression of activity-dependent bulk endocytosis in rodent models of autism spectrum disorder.
Molecular autismMutations in the small nuclear RNA gene RNU2-2 cause a severe neurodevelopmental disorder with prominent epilepsy.
Nature geneticsGenome-wide association study of idiopathic epilepsy in the Italian Spinone dog breed.
PloS oneRare disease gene association discovery in the 100,000 Genomes Project.
NatureState-of-the-art gene therapy in epilepsy.
Current opinion in neurologyRoadmap to advance therapeutics for SYNGAP1-related disorder: a patient organization perspective from SynGAP Research Fund.
Therapeutic advances in rare diseasePolygenic risk score to the rescue of monogenic diseases? The case of epilepsy.
EBioMedicineChildhood-onset focal epilepsy and acute para-infectious encephalopathy in a patient with biallelic QARS1 variants.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyNovel PNPLA8 variants associated with primary ovarian insufficiency, tremors, cerebellar ataxia and limb weakness: a case report and literature review.
Journal of neurologyNational survey on the prevalence of single-gene aetiologies for genetic developmental and epileptic encephalopathies in Italy.
Journal of medical geneticsHuman iPSC-derived microglia sense and dampen hyperexcitability of cortical neurons carrying the epilepsy-associated SCN2A-L1342P mutation.
The Journal of neuroscience : the official journal of the Society for NeuroscienceA homozygous TARS2 variant is a novel cause of syndromic neonatal diabetes.
Diabetic medicine : a journal of the British Diabetic AssociationInvestigating the effect of polygenic background on epilepsy phenotype in 'monogenic' families.
EBioMedicineTremor-Dominant Movement Disorder in ANKRD11- Associated KBG Syndrome.
Tremor and other hyperkinetic movements (New York, N.Y.)Clinical Neurologic Features and Evaluation of PTEN Hamartoma Tumor Syndrome: A Systematic Review.
NeurologyDiscovery of a TRMT10A mutation in a case of atypical diabetes: Case report.
Diabetes & metabolismDevelopmental and epileptic encephalopathies.
Nature reviews. Disease primersPreclinical studies of gene replacement therapy for CDKL5 deficiency disorder.
Molecular therapy : the journal of the American Society of Gene TherapyEnhanced hippocampal LTP but normal NMDA receptor and AMPA receptor function in a rat model of CDKL5 deficiency disorder.
Molecular autismThe 2017 and 2022 ILAE epilepsy classification systems identify needs and opportunities in care: A paediatric hospital-based study.
Epilepsy & behavior : E&BGenome Sequencing for Diagnosing Rare Diseases.
The New England journal of medicineNeurological Diseases and Prevalence of Antineuronal Antibodies in Patients with Autoimmune Polyendocrine Syndrome Type 1 - A National Cohort Study.
Journal of clinical immunologyEpilepsies with onset during the first year of life: A prospective study on syndromes, etiologies, and outcomes.
Epilepsia openGene replacement therapies for inherited disorders of neurotransmission: Current progress in succinic semialdehyde dehydrogenase deficiency.
Journal of inherited metabolic diseaseG protein-coupled receptor (GPCR) gene variants and human genetic disease.
Critical reviews in clinical laboratory sciencesPreimplantation genetic testing for monogenic disorders (PGT-M) offers an alternative strategy to prevent children from being born with hereditary neurological diseases or metabolic diseases dominated by nervous system phenotypes: a retrospective study.
Journal of assisted reproduction and geneticsCerebral visual impairment: genetic diagnoses and phenotypic associations.
Journal of medical geneticsGenetic and phenotypic landscape of pediatric-onset epilepsy in 142 Indian families: Counseling and therapeutic implications.
Clinical geneticsGenetic architecture of childhood speech disorder: a review.
Molecular psychiatryNext-generation sequencing testing in children with epilepsy reveals novel clinical, diagnostic and therapeutic implications.
Frontiers in geneticsRare disease gene association discovery from burden analysis of the 100,000 Genomes Project data.
medRxiv : the preprint server for health sciencesFDA Patient-Focused Drug Development Guidances: Considerations for Trial Readiness in Rare Developmental and Epileptic Encephalopathies.
NeurologyDistinctive In Vitro Phenotypes in iPSC-Derived Neurons From Patients With Gain- and Loss-of-Function SCN2A Developmental and Epileptic Encephalopathy.
The Journal of neuroscience : the official journal of the Society for NeuroscienceLeveraging multiple approaches for the detection of pathogenic deep intronic variants in developmental and epileptic encephalopathies: A case report.
Epilepsia openProspective evaluation of NGS-based sequencing in epilepsy patients: results of seven NASGE-associated diagnostic laboratories.
Frontiers in neurologyThe role of copy number variants in the genetic architecture of common familial epilepsies.
EpilepsiaPentanucleotide Repeat Insertions in RAI1 Cause Benign Adult Familial Myoclonic Epilepsy Type 8.
Movement disorders : official journal of the Movement Disorder SocietyDe novo variants in RNF213 are associated with a clinical spectrum ranging from Leigh syndrome to early-onset stroke.
Genetics in medicine : official journal of the American College of Medical GeneticsBK Channelopathies and KCNMA1-Linked Disease Models.
Annual review of physiologyElectroclinical features and phenotypic differences in adenylosuccinate lyase deficiency: Long-term follow-up of seven patients from four families and appraisal of the literature.
Epilepsia openRNA therapeutics for epilepsy: An emerging modality for drug discovery.
EpilepsiaSafe use of the ketogenic diet in an infant with microcephaly, epilepsy, and diabetes syndrome: a case report.
BMC pediatricsRecurrent Ischemic Strokes due to Monogenic COL4A1 Mutation: The First Case Report from Latin America.
Case reports in geneticsReversal of cell, circuit and seizure phenotypes in a mouse model of DNM1 epileptic encephalopathy.
Nature communicationsFamilial Mesial Temporal Lobe Epilepsy: Clinical Spectrum and Genetic Evidence for a Polygenic Architecture.
Annals of neurologyDiet in treatment of autism spectrum disorders.
Frontiers in neuroscienceRecent advances in neurometabolic diseases: The genetic role in the modern era.
Epilepsy & behavior : E&BStrain-dependent effects on neurobehavioral and seizure phenotypes in Scn2aK1422E mice.
bioRxiv : the preprint server for biologyZebrafish as an Innovative Tool for Epilepsy Modeling: State of the Art and Potential Future Directions.
International journal of molecular sciencesExome Sequencing and Multigene Panel Testing in 1,411 Patients With Adult-Onset Neurologic Disorders.
Neurology. GeneticsImplementation of Exome Sequencing in Clinical Practice for Neurological Disorders.
GenesWidespread genomic influences on phenotype in Dravet syndrome, a 'monogenic' condition.
Brain : a journal of neurologyCase report: Neonatal diabetes mellitus caused by KCNJ11 mutation presenting with intracranial hemorrhage.
Frontiers in neurologyPediatric-Onset Epilepsy and Developmental Epileptic Encephalopathies Followed by Early-Onset Parkinsonism.
International journal of molecular sciencesDe Novo Variant in the KCNJ9 Gene as a Possible Cause of Neonatal Seizures.
GenesNeuropathy, Ataxia, and Retinitis Pigmentosa Syndrome.
Journal of clinical neuromuscular diseaseGenes4Epilepsy: An epilepsy gene resource.
EpilepsiaHomozygous Missense Variant in the N-Terminal Region of ANK3 Gene Is Associated with Developmental Delay, Seizures, Speech Abnormality, and Aggressive Behavior.
Molecular syndromologyCytogenomic epileptology.
Molecular cytogeneticsGRIN2A-related epilepsy and speech disorders: A comprehensive overview with a focus on the role of precision therapeutics.
Epilepsy researchTranscriptomic analyses reveal neuronal specificity of Leigh syndrome associated genes.
Journal of inherited metabolic diseaseEstimating the Prevalence of De Novo Monogenic Neurodevelopmental Disorders from Large Cohort Studies.
BiomedicinesAssociation of Vascular Risk Factors and Genetic Factors With Penetrance of Variants Causing Monogenic Stroke.
JAMA neurologyA rigorous in silico genomic interrogation at 1p13.3 reveals 16 autosomal dominant candidate genes in syndromic neurodevelopmental disorders.
Frontiers in molecular neuroscienceThe phenotypic spectrum associated with loss-of-function variants in monogenic epilepsy genes in the general population.
European journal of human genetics : EJHGA systems medicine strategy to predict the efficacy of drugs for monogenic epilepsies.
EpilepsiaSLC13A5 Deficiency Disorder: From Genetics to Gene Therapy.
GenesGABRG1 variant as a potential novel cause of epileptic encephalopathy, hypotonia, and global developmental delay.
American journal of medical genetics. Part AExpression of a Secretable, Cell-Penetrating CDKL5 Protein Enhances the Efficacy of Gene Therapy for CDKL5 Deficiency Disorder.
Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeuticsMulti-Omic Investigations of a 17-19 Translocation Links MINK1 Disruption to Autism, Epilepsy and Osteoporosis.
International journal of molecular sciencesMonogenic developmental and epileptic encephalopathies of infancy and childhood, a population cohort from Norway.
Frontiers in pediatricsGene Therapy: Novel Approaches to Targeting Monogenic Epilepsies.
Frontiers in neurologyUnderstanding Protein Protocadherin-19 (PCDH19) Syndrome: A Literature Review of the Pathophysiology.
CureusGenomics in the presurgical epilepsy evaluation.
Epilepsy researchSulfonylurea for improving neurological features in neonatal diabetes: A systematic review and meta-analyses.
Pediatric diabetesCommon risk variants for epilepsy are enriched in families previously targeted for rare monogenic variant discovery.
EBioMedicineSEMA6B variants cause intellectual disability and alter dendritic spine density and axon guidance.
Human molecular geneticsWhole exome sequencing and co-expression analysis identify an SCN1A variant that modifies pathogenicity in a family with genetic epilepsy and febrile seizures plus.
EpilepsiaMultiple Early-Life Seizures Alters Neonatal Communicative Behavior in Fmr1 Knockout Mice.
Developmental neurosciencePathophysiological Heterogeneity of the BBSOA Neurodevelopmental Syndrome.
Cells'Channeling' therapeutic discovery for epileptic encephalopathy through iPSC technologies.
Trends in pharmacological sciencesEfficacy of Ketogenic Diet for Infantile Spasms in Chinese Patients With or Without Monogenic Etiology.
Frontiers in pediatricsReflex seizures in rare monogenic epilepsies.
SeizureInhibition of MEK-ERK signaling reduces seizures in two mouse models of tuberous sclerosis complex.
Epilepsy researchThe EPIGENE network: A French initiative to harmonize and improve the nationwide diagnosis of monogenic epilepsies.
European journal of medical geneticsDevelopment and Validation of a Prediction Model for Early Diagnosis of SCN1A-Related Epilepsies.
NeurologyGenetic diagnosis of basal ganglia disease in childhood.
Developmental medicine and child neurologyStructural and Functional Aspects of the Neurodevelopmental Gene NR2F1: From Animal Models to Human Pathology.
Frontiers in molecular neuroscienceDe novo coding variants in the AGO1 gene cause a neurodevelopmental disorder with intellectual disability.
Journal of medical geneticsCase Report: Lennox-Gastaut Epileptic Encephalopathy Responsive to Cannabidiol Treatment Associated With a Novel de novo Mosaic SHANK1 Variant.
Frontiers in geneticsIdentification and quantification of oligogenic loss-of-function disorders.
Genetics in medicine : official journal of the American College of Medical GeneticsNovel treatments in epilepsy guided by genetic diagnosis.
British journal of clinical pharmacology100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care - Preliminary Report.
The New England journal of medicineMonogenic Epilepsies: Disease Mechanisms, Clinical Phenotypes, and Targeted Therapies.
NeurologyRett Syndrome Spectrum in Monogenic Developmental-Epileptic Encephalopathies and Epilepsies: A Review.
GenesCRISPRa-Mediated Upregulation of scn1laa During Early Development Causes Epileptiform Activity and dCas9-Associated Toxicity.
The CRISPR journalDystonia as a prominent presenting feature in developmental and epileptic encephalopathies: A case series.
Parkinsonism & related disordersGenotype-Phenotype Relations for the Atypical Parkinsonism Genes: MDSGene Systematic Review.
Movement disorders : official journal of the Movement Disorder SocietyExpanding the KIF4A-associated phenotype.
American journal of medical genetics. Part APredictors of cognitive, behavioural and academic difficulties in NF1.
Journal of psychiatric researchPPFIA4 mutation: A second hit in POLG related disease?
Epilepsy & behavior reportsTSPEAR variants are primarily associated with ectodermal dysplasia and tooth agenesis but not hearing loss: A novel cohort study.
American journal of medical genetics. Part ARetinoschisis and Norrie disease: a missing link.
BMC research notesElp2 mutations perturb the epitranscriptome and lead to a complex neurodevelopmental phenotype.
Nature communicationsIntegration of whole genome sequencing into a healthcare setting: high diagnostic rates across multiple clinical entities in 3219 rare disease patients.
Genome medicineAAV-Mediated Gene Therapy for Glycosphingolipid Biosynthesis Deficiencies.
Trends in molecular medicineGeneration and characterization of human induced pluripotent stem cells (iPSCs) from three male and three female patients with CDKL5 Deficiency Disorder (CDD).
Stem cell researchEtiologic distribution and clinical characteristics of pediatric diabetes in 276 children and adolescents with diabetes at a single academic center.
BMC pediatricsGenetic Factors Underlying Sudden Infant Death Syndrome.
The application of clinical geneticsExpanding the phenotype, genotype and biochemical knowledge of ALG3-CDG.
Journal of inherited metabolic diseaseGenetic Dystonias: Update on Classification and New Genetic Discoveries.
Current neurology and neuroscience reportsDe novo TRIM8 variants impair its protein localization to nuclear bodies and cause developmental delay, epilepsy, and focal segmental glomerulosclerosis.
American journal of human geneticsFBXO28 causes developmental and epileptic encephalopathy with profound intellectual disability.
EpilepsiaModelling genetic mosaicism of neurodevelopmental disorders in vivo by a Cre-amplifying fluorescent reporter.
Nature communicationsNo association between SCN9A and monogenic human epilepsy disorders.
PLoS geneticsClinical and genetic characteristics and prenatal diagnosis of patients presented GDD/ID with rare monogenic causes.
Orphanet journal of rare diseasesThe off-label use of anakinra in pediatric systemic autoinflammatory diseases.
Therapeutic advances in musculoskeletal diseaseEpilepsy and neurobehavioral abnormalities in mice with a dominant-negative KCNB1 pathogenic variant.
Neurobiology of diseaseMonogenic variants in dystonia: an exome-wide sequencing study.
The Lancet. NeurologyPrenatal diagnosis of Norrie disease after whole exome sequencing of an affected proband during an ongoing pregnancy: a case report.
BMC medical geneticsTuberous Sclerosis Complex as Disease Model for Investigating mTOR-Related Gliopathy During Epileptogenesis.
Frontiers in neurologyNeurological disorder-associated genetic variants in individuals with psychogenic nonepileptic seizures.
Scientific reportsMutations of the Transcriptional Corepressor ZMYM2 Cause Syndromic Urinary Tract Malformations.
American journal of human geneticsEarly-onset epileptic encephalopathy with migrating focal seizures associated with a FARS2 homozygous nonsense variant.
Epileptic disorders : international epilepsy journal with videotape[Hereditary Parkinson’s disease as a new clinical manifestation of the damaged POLG gene].
Orvosi hetilapExpansionHunter Denovo: a computational method for locating known and novel repeat expansions in short-read sequencing data.
Genome biologyBi-allelic GAD1 variants cause a neonatal onset syndromic developmental and epileptic encephalopathy.
Brain : a journal of neurologyCACNA1H variants are not a cause of monogenic epilepsy.
Human mutationIschemic Stroke in Young Adults.
Continuum (Minneapolis, Minn.)A deletion in Eml1 leads to bilateral subcortical heterotopia in the tish rat.
Neurobiology of diseaseA catalogue of new incidence estimates of monogenic neurodevelopmental disorders caused by de novo variants.
Brain : a journal of neurologyMigraine With Aura as Early Disease Marker in Hereditary Dutch-Type Cerebral Amyloid Angiopathy.
StrokeGrandparental genotyping enhances exome variant interpretation.
American journal of medical genetics. Part AFrom Genetic Testing to Precision Medicine in Epilepsy.
Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeuticsChromatin remodeling dysfunction extends the etiological spectrum of schizophrenia: a case report.
BMC medical geneticsStructural Noninfectious Manifestations of the Central Nervous System in Common Variable Immunodeficiency Disorders.
The journal of allergy and clinical immunology. In practiceA systems-level framework for anti-epilepsy drug discovery.
NeuropharmacologyPharmacological Treatments for Fragile X Syndrome Based on Synaptic Dysfunction.
Current pharmaceutical designDe novo NSF mutations cause early infantile epileptic encephalopathy.
Annals of clinical and translational neurologyA novel lethal recognizable polymicrogyric syndrome caused by ATP1A2 homozygous truncating variants.
Brain : a journal of neurologyImpact on Clinical Decision Making of Next-Generation Sequencing in Pediatric Epilepsy in a Tertiary Epilepsy Referral Center.
Clinical EEG and neurosciencePharmacological approaches to tackle NCLs.
Biochimica et biophysica acta. Molecular basis of diseaseNew avenues in molecular genetics for the diagnosis and application of therapeutics to the epilepsies.
Epilepsy & behavior : E&BTackling Epilepsy With High-definition Precision Medicine: A Review.
JAMA neurologyPathogenic WDFY3 variants cause neurodevelopmental disorders and opposing effects on brain size.
Brain : a journal of neurologyGenetics of neonatal-onset epilepsies.
Handbook of clinical neurologyDEGS1 variant causes neurological disorder.
European journal of human genetics : EJHGBiallelic mutations in PIGP cause developmental and epileptic encephalopathy.
Annals of clinical and translational neurologyGenetics of neonatal onset epilepsies: An overview.
Revue neurologiqueMouse Models of Familial Hemiplegic Migraine for Studying Migraine Pathophysiology.
Current neuropharmacologyDe novo SCN1A, SCN8A, and CLCN2 mutations in childhood absence epilepsy.
Epilepsy researchStiripentol protects against calcium oxalate nephrolithiasis and ethylene glycol poisoning.
The Journal of clinical investigationContribution of ultrarare variants in mTOR pathway genes to sporadic focal epilepsies.
Annals of clinical and translational neurologyNovel ANKRD11 gene mutation in an individual with a mild phenotype of KBG syndrome associated to a GEFS+ phenotypic spectrum: a case report.
BMC medical geneticsIdentification of a Loss-of-Function Mutation in the Context of Glutaminase Deficiency and Neonatal Epileptic Encephalopathy.
JAMA neurologyGenome-wide mega-analysis identifies 16 loci and highlights diverse biological mechanisms in the common epilepsies.
Nature communications[Whole Exome Sequencing in daily practice: the possibilities and impossibilities of this diagnostic test].
Nederlands tijdschrift voor geneeskundeMultiple Symmetric Lipomatosis (Madelung Disease) in a Large Canadian Family With the Mitochondrial MTTK c.8344A>G Variant.
Journal of investigative medicine high impact case reportsBlood-Brain Barrier: From Physiology to Disease and Back.
Physiological reviewsExome sequencing in adult neurology practice: Challenges and rewards in a mixed resource setting.
Clinical neurology and neurosurgeryBeta-propeller protein-associated neurodegeneration (BPAN) as a genetically simple model of multifaceted neuropathology resulting from defects in autophagy.
Reviews in the neurosciencesA novel FBXO28 frameshift mutation in a child with developmental delay, dysmorphic features, and intractable epilepsy: A second gene that may contribute to the 1q41-q42 deletion phenotype.
American journal of medical genetics. Part ATo diet or not to diet in neonatal diabetes responding to sulfonylurea treatment.
Journal of pediatric endocrinology & metabolism : JPEMIdentification of new risk factors for rolandic epilepsy: CNV at Xp22.31 and alterations at cholinergic synapses.
Journal of medical geneticsElectrocorticographic telemetric recording in unrestrained mouse pups.
Journal of neuroscience methodsExpansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy.
Nature geneticsTargeted gene panel and genotype-phenotype correlation in children with developmental and epileptic encephalopathy.
Epilepsy researchAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Doença monogênica com epilepsia.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Doença monogênica com epilepsia
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Characterizing SCN1A -Related Disorders Using Real-World Data Across 681 Patient-Years.
- Mitochondrial Dysfunction in Monogenic Developmental and Epileptic Encephalopathies.
- Research progress in SYNGAP1-related neurodevelopmental disorders: from pathogenesis to therapeutic strategies.
- International Clinical Evidence-based Guideline for Kleefstra Syndrome.Genetics in medicine : official journal of the American College of Medical Genetics· 2026· PMID 41578867mais citado
- Seizure outcomes after VNS therapy in children with drug-resistant epilepsy due to monogenic etiologies versus malformations of cortical development.
- Molecular Profiling of Polish Pediatric Patients with Epilepsy: A Single-Center Diagnostic Experience Using Next-Generation Sequencing.
- Altered Auditory Maturation in Fragile X Syndrome and Its Involvement in Audiogenic Seizure Susceptibility.
- Genetic Variants and Disease Mechanisms: Lessons from Monogenic Childhood Epilepsies.
- Clinical and genetic landscape of epilepsies with absence seizures and single-gene etiology.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:166472(Orphanet)
- MONDO:0015653(MONDO)
- Epilepsia(PCDT · Ministério da Saúde)
- GARD:20086(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55785628(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Doença monogênica com epilepsia
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Reposicionamento
- fonte: Drug Repurposing Hub