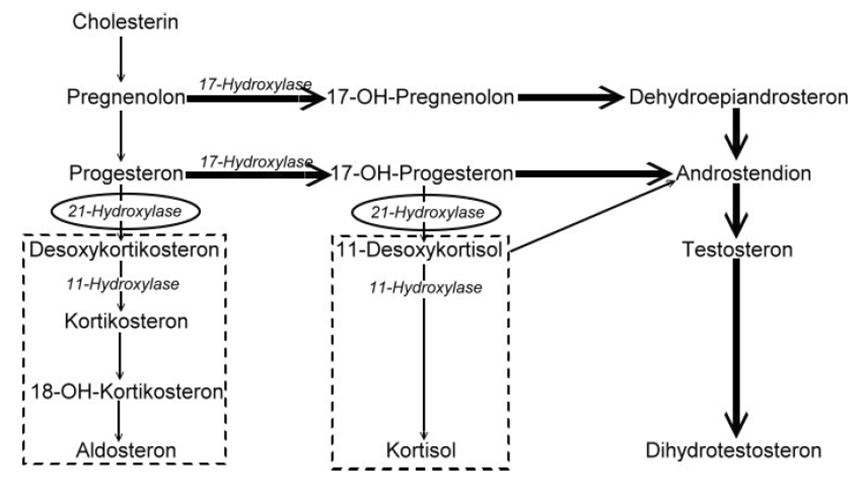
A Hiperplasia Adrenal Congênita (HAC) causada pela falta da enzima 11-beta-hidroxilase (também conhecida como CYP11B1) é uma doença genética rara que afeta as glândulas adrenais (que ficam acima dos rins e produzem hormônios). Ela se caracteriza pela falta de glicocorticoides (um tipo de hormônio essencial para o corpo), pelo excesso de andrógenos (hormônios masculinos), por pressão alta e, em meninas e mulheres, pelo desenvolvimento de características masculinas.
Introdução
O que você precisa saber de cara
A Hiperplasia Adrenal Congênita (HAC) causada pela falta da enzima 11-beta-hidroxilase (também conhecida como CYP11B1) é uma doença genética rara que afeta as glândulas adrenais (que ficam acima dos rins e produzem hormônios). Ela se caracteriza pela falta de glicocorticoides (um tipo de hormônio essencial para o corpo), pelo excesso de andrógenos (hormônios masculinos), por pressão alta e, em meninas e mulheres, pelo desenvolvimento de características masculinas.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 27 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 42 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Triagem neonatal (Teste do Pezinho)
A triagem neonatal permite diagnóstico precoce e início imediato do tratamento.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisA cytochrome P450 monooxygenase involved in the biosynthesis of adrenal corticoids (PubMed:12530636, PubMed:1518866, PubMed:1775135, PubMed:18215163, PubMed:23322723). Catalyzes a variety of reactions that are essential for many species, including detoxification, defense, and the formation of endogenous chemicals like steroid hormones. Steroid 11beta, 18- and 19-hydroxylase with preferred regioselectivity at 11beta, then 18, and lastly 19 (By similarity). Catalyzes the hydroxylation of 11-deoxyc
Mitochondrion inner membrane
Adrenal hyperplasia 4
A form of congenital adrenal hyperplasia, a common recessive disease due to defective synthesis of cortisol. Congenital adrenal hyperplasia is characterized by androgen excess leading to ambiguous genitalia in affected females, rapid somatic growth during childhood in both sexes with premature closure of the epiphyses and short adult stature. Four clinical types: 'salt wasting' (SW, the most severe type), 'simple virilizing' (SV, less severely affected patients), with normal aldosterone biosynthesis, 'non-classic form' or late-onset (NC or LOAH) and 'cryptic' (asymptomatic).
Medicamentos aprovados (FDA)
1 medicamento encontrado nos registros da FDA americana.
Variantes genéticas (ClinVar)
309 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
3 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Hiperplasia suprarrenal congênita por deficiência de 11-beta-hidroxilase
Centros de Referência SUS
24 centros habilitados pelo SUS para Hiperplasia suprarrenal congênita por deficiência de 11-beta-hidroxilase
Centros para Hiperplasia suprarrenal congênita por deficiência de 11-beta-hidroxilase
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
A Recurrent Splice Variant Sheds Light on 11β-Hydroxylase Deficiency in a Unique Large Cohort.
Congenital adrenal hyperplasia can be due to 11β-hydroxylase deficiency (11βOHD). Sporadic reports of 11βOHD are frequent but overviews on molecular landscape in some populations are lacking. The aim of this research was to compile a genetic landscape from an 11βOHD cohort, and to report a novel yet recurrent splice variant. An overview of CYP11B1 variants in a cohort of 11βOHD is presented here. The functional effect of NM_000497.4(CYP11B1):c.954 + 148C > G was studied in silico and in vitro, and a genotype-phenotype correlation study ("SPLICYP" study, No. 22_1787) was conducted. Patients with 11βOHD who underwent genetic testing at the biochemistry and molecular biology department were considered for inclusion. A total of 250 patients, diagnosed from 1990 to 2024, underwent CYP11B1 sequencing. Forty-four patients carried a novel deep intronic variant (NM_000497.4(CYP11B1):c.954 + 148C > G). Four were excluded from genotype-phenotype correlation due to missing criteria. Functional validation was performed using a Minigene Reporter Assay. We retrospectively analyzed genetic findings, clinical features of 11βOHD, and hormonal assays. The Minigene study confirmed that c.954 + 148C > G disrupts splicing by activating a cryptic donor site. Patients carrying this variant had significantly lower steroid precursor levels (P < .034) and delayed pubertal onset (P = .005) compared to severe variant carriers. This retrospective study provides genetic data in a wide cohort of 11βOHD, and identifies c.954 + 148C > G as the most recurrent variant in our Caucasian participant recruitment. Screening of deep intronic regions, coupled with functional in vitro tools, must not be overlooked in the strategy to avoid diagnostic failure.
First intragenic inversion of CYP11B1 gene causing 11β-hydroxylase deficiency: a molecular diagnosis easily overlooked.
11β-hydroxylase deficiency (11βOHD) is the second most common cause (5%) of congenital adrenal hyperplasia (CAH). The CYP11B1 gene shares 95% of genomic sequence homology with CYP11B2, and therefore Sanger sequencing remains the gold standard. We present a case of 11βOHD due to an intragenic inversion in CYP11B1 that was missed by both the Sanger sequencing and massive parallel sequencing (MPS) methods. The child was born with virilised genitalia at Prader stage 4 and the biological findings showed a hydromineral retention pattern and a pathognomonic increase in steroid precursors suggestive of 11βOHD. Standard trio analysis revealed only one heterozygous pathogenic variation inherited from the father. The study using MPS showed similar outcomes. Careful observation of the alignment BAM files revealed breaks in sequencing depth, incomplete alignments and systematic paradoxical read-pairs orientation. A specifically designed amplification and Sanger protocol confirmed the novel NM_000497.4(CYP11B1):c.[892_1121+7 inv;1121+8_1121+9del]; p.(Glu298HisfsTer113) variant at heterozygous state in the proband and his mother, fulfilling the diagnosis. The present case reports the first short intragenic inversion in CAH and illustrates the pitfalls that must always be kept in mind when using sequencing methods. When the phenotype is unequivocal, a thorough investigation of the locus should be carried out with cross-use of different techniques.
Congenital adrenal hyperplasia due to 11-beta-hydroxylase deficiency: clinical, biochemical and molecular characteristics and long-term outcomes.
11β-hydroxylase (11β-OH) deficiency is the second most frequent cause of classic congenital adrenal hyperplasia (CAH) (5%-8% of cases). Clinically, it is characterized by virilization and arterial hypertension. The objective of this study was to describe the clinical, biochemical and genetic characteristics classic 11β-OH deficiency in patients managed in our hospital and its outcomes. Retrospective longitudinal, observational and descriptive study. Patients with clinical features of virilization, high levels of 11-deoxycortisol and study of CYP11B1 gene with detection of pathogenic and likely pathogenic variants. We identified 6 patients (1 male, 5 female) from 4 families. In the 4 index cases, the median age at diagnosis was 2.3 years. The 46,XX patients exhibited a variable degree of virilization at diagnosis, with a predominance of Prader stage V, and one case of male sex assignment at birth. All patients had elevated serum concentrations of 17-hydroxyprogesterone and testosterone. Fifty percent of the patients had developed arterial hypertension during the follow-up, with onset at a median age of 9.3 years. Three 46,XX patients reached a median final height of 154 cm. Six different variants of theCYP11B1 gene were identified, 5 of which were novel variants (c.595 G > A, c.710 T > C, c.1156delG, c.395 + 2dupT, c.1159dupA). There is considerable heterogeneity in the clinical presentation of patients with CAH due to 11β-OH deficiency. Early diagnosis and treatment are important to prevent complications and improve long-term outcomes. We report 6 different variants of the CYP11B1 gene, including 5 novel variants.
[A case of 11β-hydroxylase deficiency complicated with bilateral adrenal myelolipoma].
分析1例经典型11β-羟化酶缺陷症合并双侧肾上腺髓样脂肪瘤患者的临床资料,发现1种可能致病的新突变c.600C>A(p.Ser200Arg),且侧面证实了切除双侧肾上腺对于治疗11β-羟化酶缺陷症患者难治性高血压的有效性,提高对临床罕见的11β-羟化酶缺陷症的认识。.
Clinical and molecular characterization of 10 Chinese children with congenital adrenal hyperplasia due to 11beta-hydroxylase deficiency.
The clinical manifestations of nonclassical 11beta-hydroxylase deficiency are very similar to those of non-classical 21-hydroxylase deficiency. For this study, we investigated the relationship between the clinical and molecular features of congenital adrenal hyperplasia caused by 11beta-hydroxylase deficiency and reviewed the related literature, which are expected to provide assistance for the clinical diagnosis and analysis of congenital adrenal hyperplasia. Clinical data for 10 Chinese patients diagnosed with congenital adrenal hyperplasia in our hospital from 2018 to 2022 were retrospectively analyzed. We examined the effects of gene mutations on protease activity and constructed three-dimensional structure prediction models of proteins. We describe 10 patients with 11beta-hydroxylase gene mutations (n = 5, 46,XY; n = 5, 46,XX), with 10 novel mutations were reported. Female patients received treatment at an early stage, with an average age of 2.08 ± 1.66 years, whereas male patients received treatment significantly later, at an average age of 9.77 ± 3.62 years. The most common CYP11B1 pathogenic variant in the Chinese population was found to be c.1360C > T. All mutations lead to spatial conformational changes that affect protein stability. Our study found that there was no significant correlation between each specific mutation and the severity of clinical manifestations. Different patients with the same gene pathogenic variant may have mild or severe clinical manifestations. The correlation between genotype and phenotype needs further study. Three-dimensional protein simulations may provide additional support for the physiopathological mechanism of genetic mutations.
Publicações recentes
Congenital adrenal hyperplasia due to 11-beta-hydroxylase deficiency: clinical, biochemical and molecular characteristics and long-term outcomes.
Pregnancy in a woman with congenital adrenal hyperplasia with 11-beta-hydroxylase deficiency: A case report.
Congenital adrenal hyperplasia due to 11-Beta-hydroxylase deficiency in a Tunisian family.
Clinical perspectives in congenital adrenal hyperplasia due to 11β-hydroxylase deficiency.
📚 EuropePMC28 artigos no totalmostrando 41
A Recurrent Splice Variant Sheds Light on 11β-Hydroxylase Deficiency in a Unique Large Cohort.
The Journal of clinical endocrinology and metabolismFirst intragenic inversion of CYP11B1 gene causing 11β-hydroxylase deficiency: a molecular diagnosis easily overlooked.
Journal of medical geneticsCongenital adrenal hyperplasia due to 11-beta-hydroxylase deficiency: clinical, biochemical and molecular characteristics and long-term outcomes.
Anales de pediatria[A case of 11β-hydroxylase deficiency complicated with bilateral adrenal myelolipoma].
Zhonghua nei ke za zhiTargeted long-read sequencing identifies missing pathogenic variant in unsolved 11β-hydroxylase deficiency.
BMC endocrine disordersMineralocorticoid receptor antagonist monotherapy in pediatric non-classical 11β-hydroxylase deficiency.
Journal of pediatric endocrinology & metabolism : JPEM[Analysis of a family with 11β-hydroxylase deficiency due to a mutation in the CYP11B1 gene].
Zhonghua yi xue za zhiNon-classical 11β-hydroxylase deficiency caused by a novel heterozygous mutation: a case report and review of the literature.
EndocrineClinical Presentation and Genetic Analysis of Neonatal 11β-Hydroxylase Deficiency Induced by a Chimeric CYP11B2/CYP11B1 Gene.
Journal of clinical research in pediatric endocrinologyCase Report: A combination of chimeric CYP11B2/CYP11B1 and a novel p.Val68Gly CYP11B1 variant causing 11β-Hydroxylase deficiency in a Chinese patient.
Frontiers in endocrinologyUnequal crossing over between CYP11B2 and CYP11B1 causes 11 β -hydroxylase deficiency in a consanguineous family.
The Journal of steroid biochemistry and molecular biologyClinical and molecular characterization of 10 Chinese children with congenital adrenal hyperplasia due to 11beta-hydroxylase deficiency.
World journal of pediatrics : WJPPregnancy in a woman with congenital adrenal hyperplasia with 11-beta-hydroxylase deficiency: A case report.
Obstetric medicineChallenges in the treatment of late-identified untreated congenital adrenal hyperplasia due to CYP11B1 deficiency: Lessons from a developing country.
Frontiers in endocrinologyA Novel Homozygous Pathogenic Variant in CYP11B1 in a Female Iranian Patient with 11B Hydroxylase Deficiency.
Laboratory medicineCase Report: A Novel Mutation Leading to 11-β Hydroxylase Deficiency in a Female Patient.
Endocrine, metabolic & immune disorders drug targetsAllele-specific PCR and Next-generation sequencing based genetic screening for Congenital Adrenal Hyperplasia in India.
European journal of medical geneticsCompound heterozygosity of a novel Q73X mutation and a known R141X mutation in CYP11B1 resulting in 11β-hydroxylase deficiency in a Chinese boy with congenital adrenal hyperplasia.
The Journal of steroid biochemistry and molecular biologyCongenital adrenal hyperplasia due to 11-Beta-hydroxylase deficiency in a Tunisian family.
The Pan African medical journalA Rare Cause of Virilization, Short Stature, and Hypertension.
Clinical chemistryDifferentiating 11β-hydroxylase deficiency from primary glucocorticoid resistance syndrome in male precocity: real challenge in low-income countries.
BMJ case reportsA Chinese patient with 11β-hydroxylase deficiency due to novel compound heterozygous mutation in CYP11B1 gene: a case report.
BMC endocrine disordersLong-term follow-up of a female patient with non-classical 11β-hydroxylase deficiency and two novel mutations in CYP11B1.
Gynecological endocrinology : the official journal of the International Society of Gynecological EndocrinologyTwo rare forms of congenital adrenal hyperplasia, 11β hydroxylase deficiency and 17-hydroxylase/17,20-lyase deficiency, presenting with novel mutations.
Hormones (Athens, Greece)A novel chimeric CYP11B2/CYP11B1 combined with a new p.L340P CYP11B1 mutation in a patient with 11OHD: case report.
BMC endocrine disordersPrevalence, clinical characteristics and long-term outcomes of classical 11 β-hydroxylase deficiency (11BOHD) in Turkish population and novel mutations in CYP11B1 gene.
The Journal of steroid biochemistry and molecular biologyCongenital adrenal hyperplasia causing hypertension: an illustrative review.
Journal of human hypertensionA high rate of novel CYP11B1 mutations in Saudi Arabia.
The Journal of steroid biochemistry and molecular biologyTesticular adrenal rest tumors in congenital adrenal hyperplasia-cross-sectional study of 51 Croatian male patients.
European journal of pediatricsCongenital Adrenal Hyperplasia with 11-Beta Hydroxylase Deficiency with Testicular Adrenal Rest Tumour.
The Journal of the Association of Physicians of IndiaA child with hypertension and ambiguous genitalia - an uncommon variant of congenital adrenal hyperplasia: a case report.
Journal of medical case reportsClinical, genetic, and structural basis of congenital adrenal hyperplasia due to 11β-hydroxylase deficiency.
Proceedings of the National Academy of Sciences of the United States of AmericaClinical perspectives in congenital adrenal hyperplasia due to 11β-hydroxylase deficiency.
EndocrineCongenital adrenal hyperplasia due to 11-beta-hydroxylase deficiency: description of a new mutation, R384X.
Colombia medica (Cali, Colombia)A novel CYP11B1 mutation in a Turkish patient with 11β-hydroxylase deficiency: An association with the severe hypokalemia leading to rhabdomyolysis.
Hormones (Athens, Greece)Genetic screening of non-classic CAH females with hyperandrogenemia identifies a novel CYP11B1 gene mutation.
Hormones (Athens, Greece)Long-term glucocorticoid effect on bone mineral density in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency.
European journal of endocrinologyNovel and prevalent CYP11B1 gene mutations in Turkish patients with 11-β hydroxylase deficiency.
The Journal of steroid biochemistry and molecular biologyThe combination of a novel 2 bp deletion mutation and p.D63H in CYP11B1 cause congenital adrenal hyperplasia due to steroid 11β-hydroxylase deficiency.
Endocrine journal[Change in stature after pseudo-puberty early by 11ß hydroxylase deficiency in a girl of 7 years: report of a case and review of literature].
The Pan African medical journalCharacterization of the molecular genetic pathology in patients with 11β-hydroxylase deficiency.
Clinical endocrinologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Hiperplasia suprarrenal congênita por deficiência de 11-beta-hidroxilase.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Hiperplasia suprarrenal congênita por deficiência de 11-beta-hidroxilase
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- A Recurrent Splice Variant Sheds Light on 11β-Hydroxylase Deficiency in a Unique Large Cohort.
- First intragenic inversion of CYP11B1 gene causing 11β-hydroxylase deficiency: a molecular diagnosis easily overlooked.
- Congenital adrenal hyperplasia due to 11-beta-hydroxylase deficiency: clinical, biochemical and molecular characteristics and long-term outcomes.
- [A case of 11β-hydroxylase deficiency complicated with bilateral adrenal myelolipoma].
- Clinical and molecular characterization of 10 Chinese children with congenital adrenal hyperplasia due to 11beta-hydroxylase deficiency.
- Pregnancy in a woman with congenital adrenal hyperplasia with 11-beta-hydroxylase deficiency: A case report.
- Congenital adrenal hyperplasia due to 11-Beta-hydroxylase deficiency in a Tunisian family.
- Correction: Congenital adrenal hyperplasia due to 11-beta-hydroxylase deficiency: functional consequences of four CYP11B1 mutations.
- Clinical perspectives in congenital adrenal hyperplasia due to 11β-hydroxylase deficiency.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:90795(Orphanet)
- OMIM OMIM:202010(OMIM)
- MONDO:0008729(MONDO)
- GARD:5658(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q4127183(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Hiperplasia suprarrenal congênita por deficiência de 11-beta-hidroxilase
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos aprovados FDA
- fonte: FDA OpenFDA
- Reposicionamento
- fonte: Drug Repurposing Hub