A hiperplasia adrenal congênita devido à deficiência da oxidoredutase do citocromo P450 é uma forma única de hiperplasia adrenal congênita (HAC) caracterizada por deficiência de glicocorticóides, ambiguidade sexual grave em ambos os sexos e malformações esqueléticas (especialmente craniofaciais).
Introdução
O que você precisa saber de cara
A hiperplasia adrenal congênita devido à deficiência da oxidoredutase do citocromo P450 é uma forma única de hiperplasia adrenal congênita (HAC) caracterizada por deficiência de glicocorticóides, ambiguidade sexual grave em ambos os sexos e malformações esqueléticas (especialmente craniofaciais).
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 59 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 110 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Triagem neonatal (Teste do Pezinho)
A triagem neonatal permite diagnóstico precoce e início imediato do tratamento.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisThis enzyme is required for electron transfer from NADP to cytochrome P450 in microsomes. It can also provide electron transfer to heme oxygenase and cytochrome B5
Endoplasmic reticulum membrane
Antley-Bixler syndrome, with genital anomalies and disordered steroidogenesis
A disease characterized by the association of Antley-Bixler syndrome with steroidogenesis defects and abnormal genitalia. Antley-Bixler syndrome is characterized by craniosynostosis, radiohumeral synostosis present from the perinatal period, midface hypoplasia, choanal stenosis or atresia, femoral bowing and multiple joint contractures.
Medicamentos aprovados (FDA)
1 medicamento encontrado nos registros da FDA americana.
Variantes genéticas (ClinVar)
194 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 802 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Hiperplasia suprarrenal congênita por deficiência de citocromo P450 oxidorreductase
Centros de Referência SUS
24 centros habilitados pelo SUS para Hiperplasia suprarrenal congênita por deficiência de citocromo P450 oxidorreductase
Centros para Hiperplasia suprarrenal congênita por deficiência de citocromo P450 oxidorreductase
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
Pesquisa e ensaios clínicos
1 ensaios clínicos encontrados.
Publicações mais relevantes
Live Births Following IVF-FET in Two Adult Sisters with Nonclassic P450 Oxidoreductase Deficiency: A Case Report Identifying a Novel POR Variant.
Cytochrome P450 oxidoreductase deficiency (PORD) is an exceptionally rare form of congenital adrenal hyperplasia (CAH) characterized by impaired activity of multiple microsomal cytochrome P450 enzymes. In adult women, PORD frequently presents with nonspecific reproductive manifestations such as menstrual irregularities, infertility, and ovarian cysts, often mimicking polycystic ovary syndrome (PCOS) or premature ovarian insufficiency (POI). To date, successful pregnancies in affected women remain extremely rare. We describe two biological sisters with compound heterozygous POR variants c.1811A>G (p.Tyr604Cys) and c.1952_1966del (p.Gly651_His655del), both presenting with infertility and recurrent ovarian cysts but initially misdiagnosed as PCOS or POI. Both exhibited elevated serum progesterone and 17-hydroxyprogesterone (17-OHP) without overt androgen excess, consistent with the paradoxical hormonal signature of PORD. The elder sister underwent a progestin-primed ovarian stimulation (PPOS) protocol, while the younger received a short GnRH agonist protocol; in both cases, a freeze-all strategy was adopted due to supraphysiologic progesterone levels. Subsequent hormone replacement therapy frozen embryo transfer (HRT-FET) combined with glucocorticoid supplementation resulted in singleton live births in both patients. The younger sister developed preeclampsia requiring preterm cesarean delivery, highlighting potential obstetric risks. These cases represent the first report of two siblings with nonclassic PORD achieving live births through IVF-FET. Moreover, we identified a previously unreported POR variant, c.1952_1966del, p.Gly651_His655del, which expands the known mutational spectrum of PORD in the Chinese population. Our findings highlight the importance of early genetic testing in women with atypical infertility, recognition of the distinctive hormonal profile of PORD, and the value of glucocorticoid-supported artificial-cycle frozen embryo transfer as an effective reproductive strategy.
Hemodynamic and microvascular abnormalities in P450 oxidoreductase deficiency: evidence for COX-dependent dysfunction.
Cytochrome P450 oxidoreductase deficiency (PORD) is a rare form of congenital adrenal hyperplasia caused by impaired electron transfer to multiple microsomal enzymes. Hypertension in PORD has been attributed to mineralocorticoid excess; however, experimental evidence suggests that POR may also regulate endothelial vasodilatory pathways. The vascular effects of PORD in humans remain poorly defined. We evaluated blood pressure regulation, microvascular structure, and serum vasoactive mediators in patients with PORD. Observational, cross-sectional study. Ten patients with genetically confirmed PORD (4 females, 6 males; age range 8-27 years) and 41 age- and sex-matched healthy controls were evaluated. Office and 24-hour ambulatory blood pressure monitoring (ABPM) were performed. Microvascular structure was assessed using nailfold capillaroscopy (NFC). Serum levels of thromboxane B₂ (TXB₂), prostaglandin E₂ (PGE₂), and nitric oxide (NO) were measured. Hypertension was present in 80% of patients with PORD compared with 7.3% of controls (P < .001). Office and ambulatory systolic and diastolic pressures were significantly higher in PORD. NFC revealed shorter capillary length (P = .001), more frequent crossed capillaries (P < .001), and avascular areas (P = .039). Serum TXB₂ levels were higher in PORD (P = .007), whereas PGE₂ and NO did not differ between groups. PORD is associated with an increased prevalence of hypertension, microvascular capillary abnormalities, and elevated TXB₂, consistent with enhanced cyclooxygenase-mediated vasoconstrictor tone and endothelial dysfunction. These findings suggest that cyclooxygenase-dependent mechanisms may contribute to the vascular alterations observed in PORD.
Clinical Characteristics and Molecular Aetiology of Cytochrome P450 Oxidoreductase Deficiency Diagnosed in 46,XX Patients.
P450 oxidoreductase deficiency (PORD) affects cytochrome enzyme activities, causing various symptoms, such as adrenal insufficiency, disorders of sex development and skeletal malformations. This study aims to elucidate the clinical manifestations, genotype characteristics, diagnosis and management of 46,XX karyotype patients with PORD in China. A retrospective study included twelve 46,XX PORD patients in a Chinese tertiary medical center from 2004 to 2024. The patients' clinical characteristics were summarized based on manifestations, hormone profiles, and responses to treatments. The age of first visit was 7-31 years. Except for one young girl presenting with ambiguous genitalia since born, 11 patients presented with either abnormal menses or multiple ovarian cysts. Six patients showed masculinization of their external genitalia, and ten patients showed varying degrees of skeletal deformity. Progesterone was elevated and ovarian reserve was poor in all patients. The most frequent POR variant, c.1370G > A, located in exon 11 occurred in 11/12 patients with an allele frequency of 87.5% (21/24). Two novel nonsense mutations, c.1684dupG and c.2040dupC, were identified and assessed as pathogenic and likely pathogenic by ACMG, respectively. The c.1370G > A might be a dominant mutation type of POR in China. Female patients with PORD have a vulnerable ovarian reserve, and their ovarian macrocysts can be managed conservatively for fertility preservation. This study specifically focuses on PORD in 46,XX Chinese individuals, which implies its genetic causes with novel genetic findings and summarizes the puzzling spectrum of clinical manifestations.
Measurement of twenty-one serum steroid profiles by UPLC-MS/MS for the diagnosis and monitoring of congenital adrenal hyperplasia.
Congenital adrenal hyperplasia (CAH) represents a group of inherited disorders affecting steroidogenesis. Early and accurate diagnosis is crucial for effective treatment, particularly for preventing adrenal insufficiency and minimizing androgen excess. This study aims to develop and validate an ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) method for the simultaneous quantification of 21 steroid hormones, including 11-oxygenated androgens, which are critical for diagnosing and monitoring various forms of CAH. We utilized a microbore column UPLC combined with hydroxylamine derivatization, which enabled excellent chromatographic separation and enhanced sensitivity of all target compounds. The method was evaluated for precision, linearity, recovery, ion suppression, and carryover according to FDA and CLSI guidelines. Steroid profiles from healthy controls and CAH patients were compared using Mann-Whitney tests. The UPLC-MS/MS method demonstrated excellent precision (<20 % except for 11-ketoandrostenedione), linearity (R 2 > 0.99), low limits of detection and quantification, and satisfactory recovery (57-86 % absolute, 99-111 % relative). Our method showed good correlation with proficiency testing group means, although significant negative biases were noted for androstenedione, progesterone, and 11-deoxycortisol. In a clinical setting, significant increases in pregnenolone, progesterone, 17-hydroxyprogesterone, dehydroepiandrosterone, and other key steroids were observed in patients with 21-hydroxylase deficiency, while distinct profiles were identified for patients with 17-hydroxylase deficiency, cytochrome P450 oxidoreductase deficiency, and lipoid CAH. Our UPLC-MS/MS method provides a sensitive and specific tool for the comprehensive profiling of adrenal steroids, offering improved diagnostic accuracy for CAH. Its ability to differentiate between various CAH subtypes highlights its potential clinical utility in both diagnosis and monitoring.
The distinctive P450 oxidoreductase (PORD) urinary steroid metabolome in the first week of life: Report of three cases with severe disorder.
P450 oxidoreductase (POR) facilitates electron flux to type 2 microsomal P450 cytochrome enzymes (CYPs), including the adrenal steroidogenic enzymes CYP17A1 and CYP21A2. Due to the combined impairment of these enzymes, POR deficiency (PORD), an autosomal recessive condition, results in congenital adrenal hyperplasia characterised by combined glucocorticoid and postnatal sex steroid deficiency. This study focuses on urinary steroid excretion in infants affected by PORD in the first week of life. We report on three neonatal PORD cases from two families. One family had two affected babies born three years apart who were stillborn and first-day deceased, respectively. DNA sequencing revealed a homozygous 3 bp deletion in exon six leading to an glutamic acid deletion (p.[Glu217del]). Bladder contents were obtained from the stillborn baby, and excreted urine was obtained from the second baby. In a second family, their second affected newborn, antenatally diagnosed carrying the common homozygous p.(Ala287Pro) mutation, had urine collected daily during the first week of life. Steroid excretions were quantified by gas chromatography-mass spectrometry (GC-MS). The birth-day excretions were very similar in all babies. Most notable and unusual was a large excretion of unmetabolised corticosterone, suggesting inhibited catabolism to allow maximum active gluco- and mineralocorticoid availability at birth. Because CYP3A7 (16α-hydroxylase) requires POR, there was an almost complete absence of usually dominant 3β-hydroxy-Δ5 steroids (16α-OH-DHEA and 16α-OH-pregnenolone) and the usually characteristic precursor pregnenolone metabolite 5-pregnene-3β,20α-diol (pregnenediol, 5PD). In the baby sequentially studied over a week, we observed gradual maturation to the typical and familiar PORD neonatal metabolome. At the end of the period, the minimally catabolised corticosterone had diminished, and steroid excretion was completely dominated by 5PD, excreted as both mono- and disulphate conjugates. Whether this metabolome is distinctive of all PORD infants, not just those with severe manifestation, is not known. On the first day of life, standard diagnostic markers are compromised due to fetal-placental-maternal contribution and unique neonatal steroid metabolism. However, the Day 1 PORD steroid metabolome remains distinctive, and we propose using additional biochemical markers reflective of the near complete reduction of POR-dependent CYP3A7 (16α-hydroxylase) activity to improve diagnostic yield.
Publicações recentes
Clinical Characteristics and Molecular Aetiology of Cytochrome P450 Oxidoreductase Deficiency Diagnosed in 46,XX Patients.
Diagnostic challenges and management advances in cytochrome P450 oxidoreductase deficiency, a rare form of congenital adrenal hyperplasia, with 46, XX karyotype.
Congenital adrenal hyperplasia due to P450 oxidoreductase deficiency.
In Silico Analysis of PORD Mutations on the 3D Structure of P450 Oxidoreductase.
A spectrum of recessiveness among Mendelian disease variants in UK Biobank.
📚 EuropePMCmostrando 57
Live Births Following IVF-FET in Two Adult Sisters with Nonclassic P450 Oxidoreductase Deficiency: A Case Report Identifying a Novel POR Variant.
International journal of women's healthHemodynamic and microvascular abnormalities in P450 oxidoreductase deficiency: evidence for COX-dependent dysfunction.
European journal of endocrinologyMeasurement of twenty-one serum steroid profiles by UPLC-MS/MS for the diagnosis and monitoring of congenital adrenal hyperplasia.
Journal of mass spectrometry and advances in the clinical labClinical Characteristics and Molecular Aetiology of Cytochrome P450 Oxidoreductase Deficiency Diagnosed in 46,XX Patients.
Reproductive sciences (Thousand Oaks, Calif.)The distinctive P450 oxidoreductase (PORD) urinary steroid metabolome in the first week of life: Report of three cases with severe disorder.
The Journal of steroid biochemistry and molecular biologyDisordered Electron Transfer: New Forms of Defective Steroidogenesis and Mitochondriopathy.
The Journal of clinical endocrinology and metabolismCongenital Adrenal Hyperplasia with Combined 21-hydroxylase deficiency and 17α-hydroxylase/17,20-lyase deficiency: An undervirilized male.
European journal of medical geneticsExploring Novel Variants of the Cytochrome P450 Reductase Gene (POR) from the Genome Aggregation Database by Integrating Bioinformatic Tools and Functional Assays.
BiomoleculesDiagnostic challenges and management advances in cytochrome P450 oxidoreductase deficiency, a rare form of congenital adrenal hyperplasia, with 46, XX karyotype.
Frontiers in endocrinologyCongenital adrenal hyperplasia due to P450 oxidoreductase deficiency.
Frontiers in endocrinologyGenetic Testing for a Patient with Suspected 3 Beta-Hydroxysteroid Dehydrogenase Deficiency: A Case of Unreported Genetic Variants.
Journal of clinical medicineGetting pregnant with congenital adrenal hyperplasia: Assisted reproduction and pregnancy complications. A systematic review and meta-analysis.
Frontiers in endocrinologyIn Silico Analysis of PORD Mutations on the 3D Structure of P450 Oxidoreductase.
Molecules (Basel, Switzerland)A spectrum of recessiveness among Mendelian disease variants in UK Biobank.
American journal of human geneticsThe first adult case of cytochrome P450 oxidoreductase deficiency with sufficient semen volume and sperm concentration.
Congenital anomaliesClassic and current concepts in adrenal steroidogenesis: a reappraisal.
Archives of endocrinology and metabolismSteroidogenic electron-transfer factors and their diseases.
Annals of pediatric endocrinology & metabolismTwo cases of cytochrome P450 oxidoreductase deficiency with severe scoliosis and surgery requirement.
Congenital anomaliesSuccessful Delivery in 17,20-Lyase Deficiency.
The Journal of clinical endocrinology and metabolismPOR polymorphisms are associated with 21 hydroxylase deficiency.
Journal of endocrinological investigationEstablishment of an induced pluripotent stem cell line from a Antley-Bixler Syndrome (ABS) patient with the homozygote mutation p.R457H (c.1370G>A) in POR gene.
Stem cell researchLow-birth-weight infant with Antley-Bixler syndrome-like phenotype caused by POR mutation: a rare case report.
European review for medical and pharmacological sciencesCytochrome P450 oxidoreductase deficiency caused by a novel mutation in the POR gene in two siblings: case report and literature review.
Hormones (Athens, Greece)Combined homozygous 21 hydroxylase with heterozygous P450 oxidoreductase mutation in a Saudi boy presented with hypertension.
BMJ case reportsIdentifying the Misshapen Head: Craniosynostosis and Related Disorders.
Pediatrics[Clinical and genetic analysis of a pedigree affected with cytochrome P450 oxidoreductase deficiency].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsClinical and genetic analysis of cytochrome P450 oxidoreductase (POR) deficiency in a female and the analysis of a novel POR intron mutation causing alternative mRNA splicing : Overall analysis of a female with POR deficiency.
Journal of assisted reproduction and geneticsClinical characteristics of cytochrome P450 oxidoreductase deficiency: a nationwide survey in Japan.
Endocrine journalNon-classic cytochrome P450 oxidoreductase deficiency strongly linked with menstrual cycle disorders and female infertility as primary manifestations.
Human reproduction (Oxford, England)Molecular Basis of CYP19A1 Deficiency in a 46,XX Patient With R550W Mutation in POR: Expanding the PORD Phenotype.
The Journal of clinical endocrinology and metabolismNovel phenotypes and genotypes in Antley-Bixler syndrome caused by cytochrome P450 oxidoreductase deficiency: based on the first cohort of Chinese children.
Orphanet journal of rare diseasesP450 Oxidoreductase Deficiency: A Systematic Review and Meta-analysis of Genotypes, Phenotypes, and Their Relationships.
The Journal of clinical endocrinology and metabolismDifferential effects of variations in human P450 oxidoreductase on the aromatase activity of CYP19A1 polymorphisms R264C and R264H.
The Journal of steroid biochemistry and molecular biologyAlternative pathway androgen biosynthesis and human fetal female virilization.
Proceedings of the National Academy of Sciences of the United States of America[A case of Antley-Bixler syndrome caused by novel POR mutations].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsFurther Defining the Phenotypic Spectrum of B3GAT3 Mutations and Literature Review on Linkeropathy Syndromes.
GenesVariability in human drug metabolizing cytochrome P450 CYP2C9, CYP2C19 and CYP3A5 activities caused by genetic variations in cytochrome P450 oxidoreductase.
Biochemical and biophysical research communicationsPerinasal Osteotomy With Distraction Osteogenesis for a Mild Syndromic Craniosynostosis.
The Journal of craniofacial surgery[Advance in clinical research on Antley-Bixler syndrome].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsLongitudinal serum and urine steroid metabolite profiling in a 46,XY infant with prenatally identified POR deficiency.
The Journal of steroid biochemistry and molecular biologyIn vitro fertilization-frozen embryo transfer in a patient with cytochrome P450 oxidoreductase deficiency: a case report.
Gynecological endocrinology : the official journal of the International Society of Gynecological EndocrinologyA Case of Antley-Bixler Syndrome With a Novel Likely Pathogenic Variant (c.529G>C) in the POR Gene.
Annals of laboratory medicineB3GAT3-related disorder with craniosynostosis and bone fragility due to a unique mutation.
Genetics in medicine : official journal of the American College of Medical GeneticsCytochrome P450 oxidoreductase deficiency caused by R457H mutation in POR gene in Chinese: case report and literature review.
Journal of ovarian researchLong-term follow-up of a female with congenital adrenal hyperplasia due to P450-oxidoreductase deficiency.
Archives of endocrinology and metabolismP450 Oxidoreductase Deficiency: Loss of Activity Caused by Protein Instability From a Novel L374H Mutation.
The Journal of clinical endocrinology and metabolismInstability of the Human Cytochrome P450 Reductase A287P Variant Is the Major Contributor to Its Antley-Bixler Syndrome-like Phenotype.
The Journal of biological chemistryBiallelic mutations in CYP26B1: A differential diagnosis for Pfeiffer and Antley-Bixler syndromes.
American journal of medical genetics. Part ADelayed diagnosis of disorder of sex development (DSD) due to P450 oxidoreductase (POR) deficiency.
Hormones (Athens, Greece)P450 Oxidoreductase deficiency: Analysis of mutations and polymorphisms.
The Journal of steroid biochemistry and molecular biologyCompound heterozygosity of a paternal submicroscopic deletion and a maternal missense mutation in POR gene: Antley-bixler syndrome phenotype in three sibling fetuses.
Birth defects research. Part A, Clinical and molecular teratologySteroid 17-hydroxylase and 17,20-lyase deficiencies, genetic and pharmacologic.
The Journal of steroid biochemistry and molecular biologyMultidisciplinary Treatment of Antley-Bixler Syndrome.
The Cleft palate-craniofacial journal : official publication of the American Cleft Palate-Craniofacial AssociationP450 oxidoreductase deficiency with maternal virilization during pregnancy.
Clinical and experimental obstetrics & gynecologyPrenatal Diagnosis of Antley-Bixler Syndrome and POR Deficiency.
The American journal of case reportsGross Motor Function of a Child With Antley-Bixler Syndrome.
Pediatric physical therapy : the official publication of the Section on Pediatrics of the American Physical Therapy AssociationFibroblast growth factor receptor signaling in kidney and lower urinary tract development.
Pediatric nephrology (Berlin, Germany)Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Hiperplasia suprarrenal congênita por deficiência de citocromo P450 oxidorreductase.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Hiperplasia suprarrenal congênita por deficiência de citocromo P450 oxidorreductase
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Live Births Following IVF-FET in Two Adult Sisters with Nonclassic P450 Oxidoreductase Deficiency: A Case Report Identifying a Novel POR Variant.
- Hemodynamic and microvascular abnormalities in P450 oxidoreductase deficiency: evidence for COX-dependent dysfunction.
- Clinical Characteristics and Molecular Aetiology of Cytochrome P450 Oxidoreductase Deficiency Diagnosed in 46,XX Patients.
- Measurement of twenty-one serum steroid profiles by UPLC-MS/MS for the diagnosis and monitoring of congenital adrenal hyperplasia.
- The distinctive P450 oxidoreductase (PORD) urinary steroid metabolome in the first week of life: Report of three cases with severe disorder.
- Diagnostic challenges and management advances in cytochrome P450 oxidoreductase deficiency, a rare form of congenital adrenal hyperplasia, with 46, XX karyotype.
- Congenital adrenal hyperplasia due to P450 oxidoreductase deficiency.
- In Silico Analysis of PORD Mutations on the 3D Structure of P450 Oxidoreductase.
- A spectrum of recessiveness among Mendelian disease variants in UK Biobank.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:95699(Orphanet)
- OMIM OMIM:613571(OMIM)
- MONDO:0013310(MONDO)
- GARD:12664(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q16740582(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
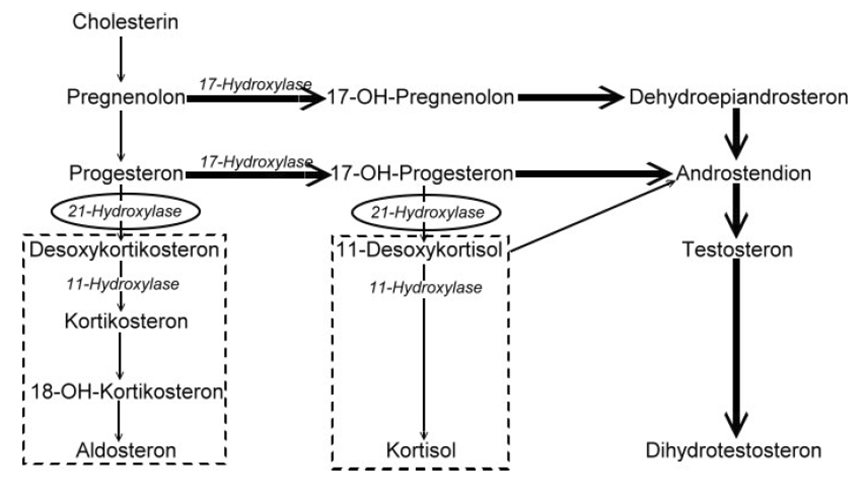
Hiperplasia suprarrenal congênita por deficiência de citocromo P450 oxidorreductase
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos aprovados FDA
- fonte: FDA OpenFDA
- Reposicionamento
- fonte: Drug Repurposing Hub