Um grupo raro e diverso de doenças metabólicas, que se caracterizam por um problema no controle do cálcio no corpo. A causa é a produção insuficiente do hormônio paratormônio (PTH). Essas condições não estão associadas a outros problemas hormonais ou defeitos de formação.
Introdução
O que você precisa saber de cara
Um grupo raro e diverso de doenças metabólicas, que se caracterizam por um problema no controle do cálcio no corpo. A causa é a produção insuficiente do hormônio paratormônio (PTH). Essas condições não estão associadas a outros problemas hormonais ou defeitos de formação.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 22 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 62 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
5 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive, X-linked recessive.
G-protein-coupled receptor that senses changes in the extracellular concentration of calcium ions and plays a key role in maintaining calcium homeostasis (PubMed:17555508, PubMed:19789209, PubMed:21566075, PubMed:22114145, PubMed:22789683, PubMed:23966241, PubMed:25104082, PubMed:25292184, PubMed:25766501, PubMed:26386835, PubMed:32817431, PubMed:33603117, PubMed:34194040, PubMed:34467854, PubMed:7759551, PubMed:8636323, PubMed:8702647, PubMed:8878438). Senses fluctuations in the circulating cal
Cell membrane
Hypocalciuric hypercalcemia, familial 1
A form of hypocalciuric hypercalcemia, a disorder of mineral homeostasis that is transmitted as an autosomal dominant trait with a high degree of penetrance. It is characterized biochemically by lifelong elevation of serum calcium concentrations and is associated with inappropriately low urinary calcium excretion and a normal or mildly elevated circulating parathyroid hormone level. Hypermagnesemia is typically present. Affected individuals are usually asymptomatic and the disorder is considered benign. However, chondrocalcinosis and pancreatitis occur in some adults.
Transcription factor playing an essential role to promote self-tolerance in the thymus by regulating the expression of a wide array of self-antigens that have the commonality of being tissue-restricted in their expression pattern in the periphery, called tissue restricted antigens (TRA) (PubMed:26084028). Binds to G-doublets in an A/T-rich environment; the preferred motif is a tandem repeat of 5'-ATTGGTTA-3' combined with a 5'-TTATTA-3' box. Binds to nucleosomes (By similarity). Binds to chromat
NucleusCytoplasm
Autoimmune polyendocrine syndrome 1, with or without reversible metaphyseal dysplasia
A rare disease characterized by the combination of chronic mucocutaneous candidiasis, hypoparathyroidism and Addison disease. Symptoms of mucocutaneous candidiasis manifest first, followed by hypotension or fatigue occurring as a result of Addison disease. APS1 is associated with other autoimmune disorders including diabetes mellitus, vitiligo, alopecia, hepatitis, pernicious anemia and primary hypothyroidism.
Guanine nucleotide-binding proteins (G proteins) function as transducers downstream of G protein-coupled receptors (GPCRs) in numerous signaling cascades (PubMed:31073061). The alpha chain contains the guanine nucleotide binding site and alternates between an active, GTP-bound state and an inactive, GDP-bound state (PubMed:31073061). Signaling by an activated GPCR promotes GDP release and GTP binding (PubMed:31073061). The alpha subunit has a low GTPase activity that converts bound GTP to GDP, t
Cell membraneCytoplasm
Hypocalciuric hypercalcemia, familial 2
A form of hypocalciuric hypercalcemia, a disorder of mineral homeostasis that is transmitted as an autosomal dominant trait with a high degree of penetrance. It is characterized biochemically by lifelong elevation of serum calcium concentrations and is associated with inappropriately low urinary calcium excretion and a normal or mildly elevated circulating parathyroid hormone level. Hypermagnesemia is typically present. Affected individuals are usually asymptomatic and the disorder is considered benign. However, chondrocalcinosis and pancreatitis occur in some adults.
Parathyroid hormone elevates calcium level by dissolving the salts in bone and preventing their renal excretion (PubMed:11604398, PubMed:35932760). Acts by binding to its receptor, PTH1R, activating G protein-coupled receptor signaling (PubMed:18375760, PubMed:35932760). Stimulates [1-14C]-2-deoxy-D-glucose (2DG) transport and glycogen synthesis in osteoblastic cells (PubMed:21076856)
Secreted
Hypoparathyroidism, familial isolated, 1
A form of hypoparathyroidism, a disorder characterized by hypocalcemia and hyperphosphatemia due to a deficiency of parathyroid hormone. Clinical features include seizures, tetany and cramps. FIH1 inheritance can be autosomal dominant or recessive.
Transcription factor that binds specific sequences on gene promoters and activate their transcription. Through the regulation of gene transcription, may play a role in parathyroid gland development
Nucleus
Hypoparathyroidism, familial isolated, 2
An autosomal recessive form of hypoparathyroidism, a disorder characterized by hypocalcemia and hyperphosphatemia due to a deficiency of parathyroid hormone. Clinical features include seizures, tetany and cramps.
Variantes genéticas (ClinVar)
948 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
15 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Hipoparatireoidismo familiar isolado
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Two novel rare variants in the PTH gene found in patients with hypoparathyroidism.
Hypoparathyroidism (HP) is a rare endocrine disorder caused by parathyroid hormone (PTH) deficiency. The PTH is a candidate gene for familial isolated hypoparathyroidism (FIH). This study aimed to investigate the pathogenicity of two novel rare variants (RVs) of PTH through in vitro functional study. Targeted next-generation sequencing was used to identify candidate gene mutations. Clinical data were retrospectively collected. Wild-type (WT) PTH was used as a template for site-directed mutagenesis to create mutant eukaryotic expression plasmids, which were transfected into cells. Treated with or without 4-phenylbutyric acid (4-PBA), the levels of intact PTH (iPTH) and PTH (1-84) were measured by chemiluminescence, and protein expression was assessed using Western blotting. Two patients carrying PTH mutations (c.154G > A: p.Val52Ile, c.270G > T: p.Leu90Phe) were identified. Patient 1, a 45-year-old male, presented with carpal and pedal numbness, muscle cramps, and low serum calcium (1.29 mmol/L). Patient 2, a 12-year-old female, had muscle twitches, convulsions, low calcium (1.50 mmol/L), and iPTH of 4 pg/mL. The iPTH or PTH (1-84) levels in the medium transfected with mutant Val52Ile and Leu90Phe PTH decreased by 31%-38%, and 51%-96% compared to WT (allP < 0.05), which were not rescued by 4-PBA. No significant changes in intracellular PTH expression were observed. In this study, two novel RVs of PTH (Val52Ile and Leu90Phe) were identified that may impair hormone synthesis and secretion. Our study has broadened the mutation spectrum of the PTH and shed light on potential mechanisms underlying FIH.
Pediatric hypoparathyroidism: etiological and clinical evaluation in a tertiary center.
This study aims to evaluate the etiology, clinical presentation, and management of pediatric hypoparathyroidism in a tertiary center. A retrospective review was conducted on pediatric patients diagnosed with hypoparathyroidism at the Pediatric Endocrinology Clinic from March 2021 to June 2023. Data on demographic characteristics, presenting symptoms, laboratory findings, genetic analyses, and treatment outcomes were collected. A total of 56 patients (31 females) were included. The median age at diagnosis of the patients was 5.5 years (range 0.04-17 years), and the median age of symptom onset was 5 years (range 0.04-16.5 years). The etiology was genetic and idiopathic in 39 patients (70.9%), with syndromic forms, familial isolated hypoparathyroidism, and hypomagnesemia identified. DiGeorge syndrome was present in 14 patients, making it the most common syndromic form. The syndromes associated with hypomagnesemia were those with mutations in the TRMP6 and CLDN16 genes. Sixteen patients (29.1%) had acquired causes, primarily post-thyroid surgery and autoimmune conditions. Common symptoms included muscle spasms (32.7%) and seizures (21.8%). Laboratory findings revealed a median serum calcium level of 6.7 mg/dL (3.8-8.5) and median serum phosphorus level of 7.7 (4.9-12.5) mg/dL. Treatment primarily involved calcitriol [The median dose of calcitriol is 25 ng/kg/day (range: 25-50 ng/kg/day)] and calcium [The median dose of calcium gluconate is 0.7 mL/kg (range: 0.5-1 mL/kg) and oral calcium is 1000 mg (range: 700-1300 mg)] supplementation. Intravenous calcium gluconate treatment was administered to 39 (70.6%) patients, oral calcium carbonate therapy was given to 16 (29.1%) patients, and calcitriol treatment was initiated for 51 (91.1%) patients. Complications such as nephrocalcinosis (7, 13.7%) and hypercalciuria (7, 13.7%) were observed in some patients. This study emphasizes the significant genetic component, particularly syndromic, in pediatric hypoparathyroidism, highlighting the need for comprehensive genetic evaluation and a multidisciplinary approach for effective management, especially concerning complications. In this way, early and accurate diagnosis will reduce unnecessary tests, treatment approaches, and repeated hospital visits. Regular monitoring is essential to mitigate potential complications associated with long-term treatment.
Three Siblings With Familial Isolated Hypoparathyroidism: A Diagnostic Journey From CASR to Novel GCM2 Variant.
We report a patient who initially presented at 4 days old with hypocalcemia, hypoparathyroidism, and elevated phosphorous level. Treatment was initiated with calcitriol, calcium carbonate (CaCO3), vitamin D, and low phosphorous formula. Family history was positive for an activating calcium sensing receptor (CASR) variant (R990G) identified previously in 2 older siblings who were treated with CaCO3 and calcitriol. However, genetic studies were negative for the CASR variant in our patient. She maintained a large calcium requirement and was admitted for multiple episodes of hypocalcemia. Further investigation revealed that the CASR variant identified in the older siblings was now considered a benign, nondisease-causing variant. Whole exome sequencing on our proband revealed a homozygous pathogenic variant in the GCM2 gene (Gln392*) consistent with a molecular diagnosis of familial isolated hypoparathyroidism. Genetic studies revealed the 2 older siblings harbor the same genetic changes and parents are heterozygous carriers for this allele. Due to persistent hypocalcemia, we initiated teriparatide. She weaned off calcitriol and achieved normocalcemia on teriparatide, CaCO3, and vitamin D. Siblings transitioned to the same treatment without complications. These findings demonstrate the importance of adequate diagnostic genetic testing and the role of variant reanalysis over time in promoting accurate diagnoses.
Case report: Familial hypoparathyroidism with elevated parathyroid hormone due to an inactivating PTH mutation.
So far, only 11 PTH mutations have been described as causes of familial isolated hypoparathyroidism (FIH). In this report, we describe a family with FIH but with significant elevation of functionally inactive PTH due to a PTH mutation. We also show a positive therapeutic outcome of recombinant human PTH (teriparatide) therapy in one of the siblings who was not well controlled on large doses of calcitriol and calcium replacement therapy. The proband is a 34-year-old woman who has a history of chronic severe hypocalcemia (HypoCa) since birth. She and her three brothers (33-year-old male twins, and a 21-year-old male) were diagnosed with pseudohypoparathyroidism type 1b (PHPT 1b) based on the presence of chronic HypoCa (serum Ca 1.6-1.85 mmol/l) since birth associated with significantly elevated plasma PTH levels in the range of 310-564 pg/dl (normal range 10-65) and absence of signs of Albright hereditary osteodystrophy. WES showed no pathogenic, likely pathogenic or variants of unknown significance in any known calcium-associated genetic disorder but a bi-allelic variant in the PTH itself ((NM_000315.4:c.128G>A, p.Gly43Glu). This was confirmed by Sanger sequencing in the patient and her affected brothers. Because the patient's HypoCa was not controlled on large doses of calcitriol and calcium carbonate, a trial of teriparatide 20 mcg SC daily was started and resulted in normalization of calcium, decline in PTH levels and significant improvement in her general wellbeing. High PTH in the presence of congenital hypocalcemia is not always due to receptor or post-receptor defect and can be due to a biologically inactive mutated PTH. In such cases, treatment with teriparatide may result in stabilization of biochemical profile and improvement in quality of life.
Full-Mouth Rehabilitation of a 15-Year-Old Girl Affected by a Rare Hypoparathyroidism (Glial Cell Missing Homolog 2 Mutation): A 3-Year Follow-Up.
Familial isolated hypoparathyroidism is a rare genetic disorder due to no or low production of the parathyroid hormone, disturbing calcium and phosphate regulation. The resulting hypocalcemia may lead to dental abnormalities, such as enamel hypoplasia. The aim of this paper was to describe the full-mouth rehabilitation of a 15-year-old girl with chronic hypocalcemia due to a rare congenital hypoparathyroidism. In this patient, in the young adult dentition, conservative care was preferred. Onlays or stainless-steel crowns were performed on the posterior teeth, and direct or indirect (overlays and veneerlays) were performed on the maxillary premolars, canines, and incisors, using a digital wax-up. The mandibular incisors were bleached. The treatment clearly improved the patient's oral quality of life, with fewer sensitivities, better chewing, and aesthetic satisfaction. The difficulties were the regular monitoring and the limited compliance of the patient. Despite no clinical feedback in the literature, generalized hypomineralized/hypoplastic teeth due to hypoparathyroidism in a young patient can be treated as amelogenesis imperfecta (generalized enamel defects) with a conservative approach for medium-term satisfactory results. This study provides new insights into the management of enamel hypoplasia caused by familial isolated hypoparathyroidism, helping to improve patient outcomes in similar cases. In primary hypoparathyroidism with hypocalcemia and hyperphosphatemia, deficient parathyroid hormone (PTH) secretion most commonly occurs from surgical excision of, or damage to, the parathyroid glands. The term idiopathic hypoparathyroidism describes isolated cases when a cause is not obvious, and there is no family history. However, hypoparathyroidism is also a feature common to a variety of hereditable syndromes that may present de novo. Familial isolated hypoparathyroidism may show autosomal dominant, autosomal recessive, or X-linked inheritance. Genes involved include PTH, SOX3, CASR, GNA11 and GCM2. Parathyroid hypoplasia is a frequent feature of 22q11.2 deletion syndrome with involvement of the TBX1 gene. The Hypoparathyroidism, Nerve Deafness, and Renal Dysplasia syndrome is due to haploinsufficiency of the GATA3 gene. Antibodies against parathyroid tissue are found in isolated hypoparathyroidism or combined with other endocrine deficiencies. Antibodies against the CASR occur in type 1 autoimmune polyglandular syndrome, due to mutations of the AIRE gene, or in acquired hypoparathyroidism. Disorders characterized by end-organ resistance to PTH are described collectively by the term pseudohypoparathyroidism (PHP), and PHP1A and PHP1B are caused by maternally-inherited changes at the imprinted GNAS complex gene that encodes the Gsα protein. Deleterious mutations of the PTH1R gene show resistance to PTH and PTHrP and present as Blomstrand lethal chondrodysplasia, Eiken syndrome, endochondromatosis, and primary failure of tooth eruption. Calcium and vitamin D are the standard therapy for the management of hypoparathyroidism, with hormone replacement [recombinant human PTH(1-84)] therapy recently becoming an option. Calcilytics, PTH analogs, and orally active small molecule PTH1R agonists may, in the future, join the treatment armamentarium. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
Publicações recentes
Two novel rare variants in the PTH gene found in patients with hypoparathyroidism.
Pediatric hypoparathyroidism: etiological and clinical evaluation in a tertiary center.
Three Siblings With Familial Isolated Hypoparathyroidism: A Diagnostic Journey From CASR to Novel GCM2 Variant.
Case report: Familial hypoparathyroidism with elevated parathyroid hormone due to an inactivating PTH mutation.
Full-Mouth Rehabilitation of a 15-Year-Old Girl Affected by a Rare Hypoparathyroidism (Glial Cell Missing Homolog 2 Mutation): A 3-Year Follow-Up.
📚 EuropePMC15 artigos no totalmostrando 10
Two novel rare variants in the PTH gene found in patients with hypoparathyroidism.
Osteoporosis and sarcopeniaPediatric hypoparathyroidism: etiological and clinical evaluation in a tertiary center.
EndocrineThree Siblings With Familial Isolated Hypoparathyroidism: A Diagnostic Journey From CASR to Novel GCM2 Variant.
JCEM case reportsCase report: Familial hypoparathyroidism with elevated parathyroid hormone due to an inactivating PTH mutation.
Frontiers in endocrinologyFull-Mouth Rehabilitation of a 15-Year-Old Girl Affected by a Rare Hypoparathyroidism (Glial Cell Missing Homolog 2 Mutation): A 3-Year Follow-Up.
Dentistry journalPrevalence of the RAPGEF5 c.2624C>A and PLOD1 c.2032G>A variants associated with equine familial isolated hypoparathyroidism and fragile foal syndrome in the US Thoroughbred population (1988-2019).
Equine veterinary journalNovel PTH Gene Mutations Causing Isolated Hypoparathyroidism.
The Journal of clinical endocrinology and metabolismHomozygous missense variant of PTH (c.166C>T, p.(Arg56Cys)) as the cause of familial isolated hypoparathyroidism in a three-year-old child.
Journal of pediatric endocrinology & metabolism : JPEMA nonsense variant in Rap Guanine Nucleotide Exchange Factor 5 (RAPGEF5) is associated with equine familial isolated hypoparathyroidism in Thoroughbred foals.
PLoS geneticsAutosomal Dominant PTH Gene Signal Sequence Mutation in a Family With Familial Isolated Hypoparathyroidism.
The Journal of clinical endocrinology and metabolismAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Hipoparatireoidismo familiar isolado.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Hipoparatireoidismo familiar isolado
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Two novel rare variants in the PTH gene found in patients with hypoparathyroidism.
- Pediatric hypoparathyroidism: etiological and clinical evaluation in a tertiary center.
- Three Siblings With Familial Isolated Hypoparathyroidism: A Diagnostic Journey From CASR to Novel GCM2 Variant.
- Case report: Familial hypoparathyroidism with elevated parathyroid hormone due to an inactivating PTH mutation.
- Full-Mouth Rehabilitation of a 15-Year-Old Girl Affected by a Rare Hypoparathyroidism (Glial Cell Missing Homolog 2 Mutation): A 3-Year Follow-Up.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2238(Orphanet)
- MONDO:0016390(MONDO)
- Hipoparatireoidismo(PCDT · Ministério da Saúde)
- GARD:2910(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q1586088(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
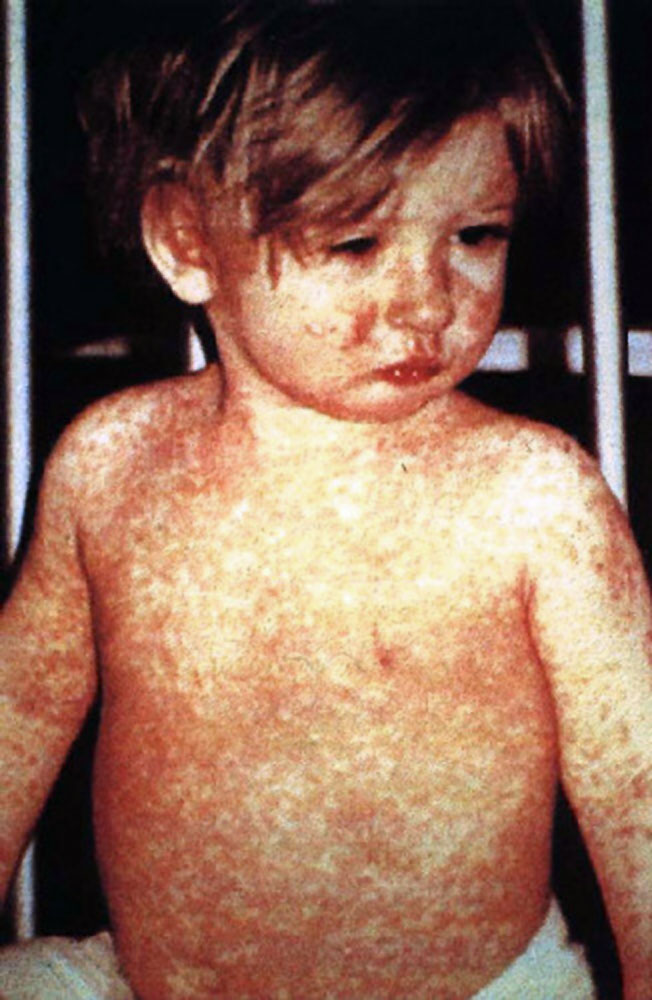
Hipoparatireoidismo familiar isolado
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Reposicionamento
- fonte: Drug Repurposing Hub