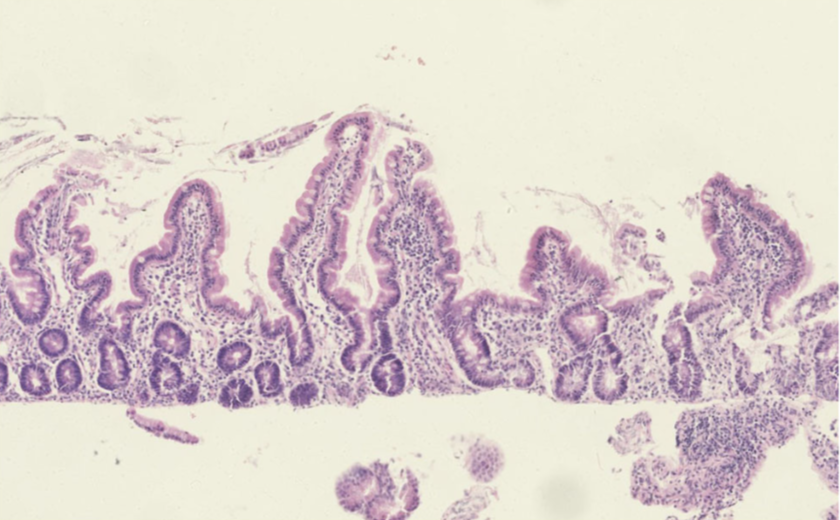
A enteropatia autoimune é uma doença autoimune rara caracterizada por perda de peso decorrente de má absorção, diarreia grave e prolongada, e dano autoimune à mucosa intestinal. A enteropatia autoimune ocorre tipicamente em lactentes e crianças pequenas; contudo, casos em adultos têm sido relatados na literatura. A enteropatia autoimune foi descrita pela primeira vez por Walker-Smith et al. em 1982.
Introdução
O que você precisa saber de cara
Visão geral
A imunodeficiência combinada por déficit de LRBA é uma doença genética rara que afeta o sistema imunológico, tornando o corpo mais vulnerável a infecções e causando inflamação em diversos órgãos. Estima-se que sua prevalência seja inferior a 1 caso por 1.000.000 de pessoas. A doença geralmente se manifesta na infância ou na primeira infância.[1][4]
Sinais e sintomas
Os principais sinais e sintomas incluem infecções respiratórias recorrentes (como pneumonia e sinusite), diarreia crônica, aumento do baço (esplenomegalia), artrite, bronquiectasia (dilatação dos brônquios), atrofia das vilosidades intestinais, hipotireoidismo, déficit de crescimento, gastrite atrófica, diabetes mellitus tipo 1, inflamação do intestino grosso, trombocitopenia (baixa contagem de plaquetas), uveíte (inflamação ocular), aumento dos gânglios linfáticos (linfadenopatia), anemia hemolítica autoimune, asma, tireoidite, vitiligo e níveis diminuídos de anticorpos (IgA, IgG e IgM).[1][4]
Causas genéticas
A doença é causada por mutações no gene LRBA (Lipopolysaccharide-responsive and beige-like anchor protein), que desempenha um papel importante na regulação do sistema imunológico. A herança é autossômica recessiva, ou seja, é necessário herdar uma cópia defeituosa do gene de cada um dos pais para desenvolver a condição.[1][2][5]
Diagnóstico
O diagnóstico é confirmado por meio de testes genéticos que identificam mutações no gene LRBA. Atualmente, existem 21 testes genéticos disponíveis e 311 variantes descritas no ClinVar. A suspeita clínica geralmente surge a partir dos sintomas característicos, como infecções recorrentes, diarreia crônica e alterações nos níveis de anticorpos.[1][2][5]
Tratamento e manejo
O manejo da imunodeficiência combinada por déficit de LRBA é focado no controle dos sintomas e na prevenção de complicações. Não há medicamentos específicos aprovados para a doença. O tratamento pode incluir reposição de imunoglobulinas, antibióticos para infecções, e cuidados para condições associadas, como diabetes ou tireoidite. É importante que o acompanhamento seja feito por uma equipe multidisciplinar. No Brasil, a doença não possui cobertura específica pelo SUS.[1][2]
Prognóstico e qualidade de vida
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
A enteropatia autoimune é uma doença autoimune rara caracterizada por perda de peso decorrente de má absorção, diarreia grave e prolongada, e dano autoimune à mucosa intestinal. A enteropatia autoimune ocorre tipicamente em lactentes e crianças pequenas; contudo, casos em adultos têm sido relatados na literatura. A enteropatia autoimune foi descrita pela primeira vez por Walker-Smith et al. em 1982.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A imunodeficiência combinada por déficit de LRBA é uma doença genética rara que afeta o sistema imunológico, tornando o corpo mais vulnerável a infecções e causando inflamação em diversos órgãos. Estima-se que sua prevalência seja inferior a 1 caso por 1.000.000 de pessoas. A doença geralmente se manifesta na infância ou na primeira infância.[1][4]
Sinais e sintomas
Os principais sinais e sintomas incluem infecções respiratórias recorrentes (como pneumonia e sinusite), diarreia crônica, aumento do baço (esplenomegalia), artrite, bronquiectasia (dilatação dos brônquios), atrofia das vilosidades intestinais, hipotireoidismo, déficit de crescimento, gastrite atrófica, diabetes mellitus tipo 1, inflamação do intestino grosso, trombocitopenia (baixa contagem de plaquetas), uveíte (inflamação ocular), aumento dos gânglios linfáticos (linfadenopatia), anemia hemolítica autoimune, asma, tireoidite, vitiligo e níveis diminuídos de anticorpos (IgA, IgG e IgM).[1][4]
Causas genéticas
A doença é causada por mutações no gene LRBA (Lipopolysaccharide-responsive and beige-like anchor protein), que desempenha um papel importante na regulação do sistema imunológico. A herança é autossômica recessiva, ou seja, é necessário herdar uma cópia defeituosa do gene de cada um dos pais para desenvolver a condição.[1][2][5]
Diagnóstico
O diagnóstico é confirmado por meio de testes genéticos que identificam mutações no gene LRBA. Atualmente, existem 21 testes genéticos disponíveis e 311 variantes descritas no ClinVar. A suspeita clínica geralmente surge a partir dos sintomas característicos, como infecções recorrentes, diarreia crônica e alterações nos níveis de anticorpos.[1][2][5]
Tratamento e manejo
O manejo da imunodeficiência combinada por déficit de LRBA é focado no controle dos sintomas e na prevenção de complicações. Não há medicamentos específicos aprovados para a doença. O tratamento pode incluir reposição de imunoglobulinas, antibióticos para infecções, e cuidados para condições associadas, como diabetes ou tireoidite. É importante que o acompanhamento seja feito por uma equipe multidisciplinar. No Brasil, a doença não possui cobertura específica pelo SUS.[1][2]
Prognóstico e qualidade de vida
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 18 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 45 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisInvolved in coupling signal transduction and vesicle trafficking to enable polarized secretion and/or membrane deposition of immune effector molecules (By similarity). Involved in phagophore growth during mitophagy by regulating ATG9A trafficking to mitochondria (PubMed:33773106)
Cell membraneEndoplasmic reticulum membraneGolgi apparatus, trans-Golgi network membraneLysosome membrane
Immunodeficiency, common variable, 8, with autoimmunity
An autosomal recessive immunologic disorder associated with defective B-cell differentiation and decreased or absent antibody production. Affected individuals have early-childhood onset of recurrent infections, particularly respiratory infections, and also develop variable autoimmune disorders, including idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and inflammatory bowel disease.
Variantes genéticas (ClinVar)
311 variantes patogênicas registradas no ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Imunodeficiência combinada por déficit de LRBA
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Imunodeficiência combinada por déficit de LRBA.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Imunodeficiência combinada por déficit de LRBA
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:445018(Orphanet)
- OMIM OMIM:614700(OMIM)
- MONDO:0013863(MONDO)
- GARD:13565(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Q25111549(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Imunodeficiência combinada por déficit de LRBA
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata