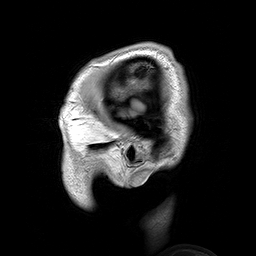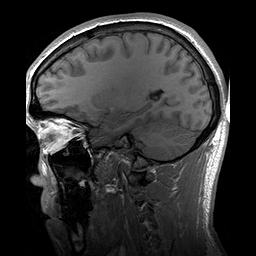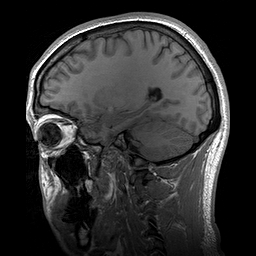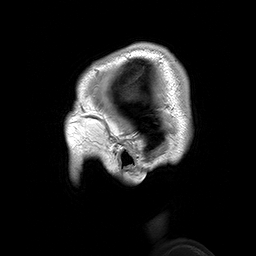
A Leucoencefalopatia Cística sem megalencefalia (que significa 'sem aumento anormal do cérebro') é uma condição caracterizada por: uma doença da substância branca do cérebro que não piora com o tempo (não progressiva); cistos (pequenas bolsas com líquido) nos dois lados da parte frontal dos lobos temporais (regiões do cérebro acima das orelhas); alterações na substância branca do cérebro; e um atraso grave no desenvolvimento das habilidades motoras e mentais (psicomotor). Até o momento, menos de 50 pacientes foram descritos na literatura médica. A forma de transmissão da doença é, muito provavelmente, autossômica recessiva.
Introdução
O que você precisa saber de cara
A Leucoencefalopatia Cística sem megalencefalia (que significa 'sem aumento anormal do cérebro') é uma condição caracterizada por: uma doença da substância branca do cérebro que não piora com o tempo (não progressiva); cistos (pequenas bolsas com líquido) nos dois lados da parte frontal dos lobos temporais (regiões do cérebro acima das orelhas); alterações na substância branca do cérebro; e um atraso grave no desenvolvimento das habilidades motoras e mentais (psicomotor). Até o momento, menos de 50 pacientes foram descritos na literatura médica. A forma de transmissão da doença é, muito provavelmente, autossômica recessiva.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 9 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 21 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisRibonuclease that plays an essential role in innate immune response by recognizing and degrading RNAs from microbial pathogens that are subsequently sensed by TLR8 (PubMed:31778653). Cleaves preferentially single-stranded RNA molecules between purine and uridine residues, which critically contributes to the supply of catabolic uridine and the generation of purine-2',3'-cyclophosphate-terminated oligoribonucleotides (PubMed:31778653, PubMed:38697119). In turn, RNase T2 degradation products promot
SecretedLysosome lumenEndoplasmic reticulum lumenMitochondrion intermembrane space
Leukoencephalopathy, cystic, without megalencephaly
An infantile-onset syndrome of cerebral leukoencephalopathy. Affected newborns develop microcephaly and neurologic abnormalities including psychomotor impairment, seizures and sensorineural hearing impairment. The brain shows multifocal white matter lesions, anterior temporal lobe subcortical cysts, pericystic abnormal myelination, ventriculomegaly and intracranial calcifications.
Accessory subunit of the mitochondrial membrane respiratory chain NADH dehydrogenase (Complex I), that is believed not to be involved in catalysis. Complex I functions in the transfer of electrons from NADH to the respiratory chain. The immediate electron acceptor for the enzyme is believed to be ubiquinone
Mitochondrion inner membrane
Mitochondrial complex I deficiency, nuclear type 13
A form of mitochondrial complex I deficiency, the most common biochemical signature of mitochondrial disorders, a group of highly heterogeneous conditions characterized by defective oxidative phosphorylation, which collectively affects 1 in 5-10000 live births. Clinical disorders have variable severity, ranging from lethal neonatal disease to adult-onset neurodegenerative disorders. Phenotypes include macrocephaly with progressive leukodystrophy, non-specific encephalopathy, cardiomyopathy, myopathy, liver disease, Leigh syndrome, Leber hereditary optic neuropathy, and some forms of Parkinson disease. MC1DN13 transmission pattern is consistent with autosomal recessive inheritance.
Variantes genéticas (ClinVar)
96 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 57 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
4 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Leucoencefalopatia cística sem megalencefalia
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Cochlear implantation in a profoundly deaf child with cystic leukoencephalopathy without megalencephaly.
Cochlear implantation candidacy criteria have continued to evolve over the years, and cochlear implantation is possible with many inner-ear and brain anomalies with good hearing and linguistic outcomes. Cystic leukoencephalopathy without megalencephaly is a rare disease in children, with only 30 cases reported in the literature, but it is associated with hearing loss in only three cases. Radiological investigations can help in diagnosing this rare entity before proceeding with cochlear implantation. A four-year-old female child born out of consanguinity with normal psychomotor development, bilateral sensorineural hearing loss and an incidental magnetic resonance imaging finding of cystic leukoencephalopathy without megalencephaly underwent successful cochlear implantation. Her post-operative period was uneventful with successful mapping of the cochlear implant. This is the first reported case of cystic leukoencephalopathy without megalencephaly and with sensorineural hearing loss in which cochlear implantation was performed successfully. White matter and temporal lobe abnormalities should not deter paediatric cochlear implantation.
Novel RNASET2 Pathogenic Variants in an East Asian Child with Delayed Psychomotor Development.
RNASET2 mutation has been reported in patients with cystic leukoencephalopathy without megalencephaly and the Aicardi-Goutieres syndrome. Both disorders are Mendelian mimics of congenital cytomegalovirus infection with overlapping features, including leukoencephalopathy, white matter alterations, intracranial calcification, delayed psychomotor development, intelligence disability and seizures. Only eight families with RNASET2 mutation have been previously reported. Whole exome sequencing was performed and copy number variants were described by read-depth strategy. We identified a novel nonsense variant c.128G>A (p. W43*) and a 430 Kb 6q27 microdeletion encompassing RNASET2. Our patient did not show anterior temporal lobe subcortical cysts, hearing loss, dystonia or extra-neurological features. Our results provided further genetic and phenotypic information of RNASET2 mutation in Chinese patients and highlighted the importance for physicians to consider RNASET2-related disorders when diagnosing patients with congenital brain infection-like phenotypes.
Clinical, radiological and possible pathological overlap of cystic leukoencephalopathy without megalencephaly and Aicardi-Goutières syndrome.
Cystic leukoencephalopathy without megalencephaly is a disorder related in some cases to RNASET2 mutations and characterized by bilateral anterior temporal subcortical cysts and multifocal lobar white matter lesions with sparing of central white matter structures. This phenotype significantly overlaps with the sequelae of in utero cytomegalovirus (CMV) infection, including the presence of intracranial calcification in some cases. Aicardi-Goutières syndrome (AGS) is another inherited leukodystrophy with cerebral calcification mimicking congenital infection. Clinical, radiological and biochemical criteria for the diagnosis of AGS have been established, although the breadth of phenotype associated with mutations in the AGS-related genes is much greater than previously envisaged. We describe the clinical, biochemical and radiological findings of five patients demonstrating a phenotype reminiscent of AGS. All patients were found to carry biallelic mutations of RNASET2. Our patients illustrate the clinical and radiological overlap that can be seen between RNASET2-related leukodystrophy and AGS in some cases. Our data highlight the need to include both disorders in the same differential diagnosis, and hint at possible shared pathomechanisms related to auto-inflammation which are worthy of further investigation.
Publicações recentes
Cochlear implantation in a profoundly deaf child with cystic leukoencephalopathy without megalencephaly.
Novel RNASET2 Pathogenic Variants in an East Asian Child with Delayed Psychomotor Development.
Clinical, radiological and possible pathological overlap of cystic leukoencephalopathy without megalencephaly and Aicardi-Goutières syndrome.
Congenital cytomegalovirus infection resembling cystic leukoencephalopathy without megalencephaly.
Cystic leukoencephalopathy without megalencephaly.
📚 EuropePMC5 artigos no totalmostrando 3
Cochlear implantation in a profoundly deaf child with cystic leukoencephalopathy without megalencephaly.
The Journal of laryngology and otologyNovel RNASET2 Pathogenic Variants in an East Asian Child with Delayed Psychomotor Development.
Fetal and pediatric pathologyClinical, radiological and possible pathological overlap of cystic leukoencephalopathy without megalencephaly and Aicardi-Goutières syndrome.
European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology SocietyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Leucoencefalopatia cística sem megalencefalia.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Leucoencefalopatia cística sem megalencefalia
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Cochlear implantation in a profoundly deaf child with cystic leukoencephalopathy without megalencephaly.
- Novel RNASET2 Pathogenic Variants in an East Asian Child with Delayed Psychomotor Development.
- Clinical, radiological and possible pathological overlap of cystic leukoencephalopathy without megalencephaly and Aicardi-Goutières syndrome.European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology Society· 2016· PMID 27091087mais citado
- Congenital cytomegalovirus infection resembling cystic leukoencephalopathy without megalencephaly.
- Cystic leukoencephalopathy without megalencephaly.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:85136(Orphanet)
- OMIM OMIM:612951(OMIM)
- MONDO:0013058(MONDO)
- GARD:13199(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55783952(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Leucoencefalopatia cística sem megalencefalia
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata