A osteogênese imperfeita tipo II é uma forma grave e geralmente fatal de osteogênese imperfeita (OI). É uma condição genética que causa ossos extremamente frágeis, com pouca massa e que quebram com muita facilidade. Bebês com o tipo II nascem com várias fraturas nas costelas e nos ossos longos (como os das pernas e braços), apresentam deformidades visíveis, têm os ossos longos mais largos, e as radiografias do crânio mostram uma densidade óssea baixa. Além disso, a parte branca dos olhos (a esclera) pode ter uma cor escura ou azulada.
Introdução
O que você precisa saber de cara
A osteogênese imperfeita tipo II é uma forma grave e geralmente fatal de osteogênese imperfeita (OI). É uma condição genética que causa ossos extremamente frágeis, com pouca massa e que quebram com muita facilidade. Bebês com o tipo II nascem com várias fraturas nas costelas e nos ossos longos (como os das pernas e braços), apresentam deformidades visíveis, têm os ossos longos mais largos, e as radiografias do crânio mostram uma densidade óssea baixa. Além disso, a parte branca dos olhos (a esclera) pode ter uma cor escura ou azulada.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 10 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 26 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
6 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive.
Curadoria gene-doença
fontes oficiaisType I collagen is a member of group I collagen (fibrillar forming collagen)
Secreted, extracellular space, extracellular matrix
Ehlers-Danlos syndrome, arthrochalasia type, 2
A form of Ehlers-Danlos syndrome, a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDSARTH2 is an autosomal dominant condition characterized by frequent congenital hip dislocation and extreme joint laxity with recurrent joint subluxations and minimal skin involvement.
Type I collagen is a member of group I collagen (fibrillar forming collagen)
Secreted, extracellular space, extracellular matrix
Caffey disease
An autosomal dominant disorder characterized by an infantile episode of massive subperiosteal new bone formation that typically involves the diaphyses of the long bones, mandible, and clavicles. The involved bones may also appear inflamed, with painful swelling and systemic fever often accompanying the illness. The bone changes usually begin before 5 months of age and resolve before 2 years of age.
PPIase that catalyzes the cis-trans isomerization of proline imidic peptide bonds in oligopeptides and may therefore assist protein folding
VirionEndoplasmic reticulum lumenMelanosome
Osteogenesis imperfecta 9
A form of osteogenesis imperfecta, a disorder of bone formation characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI9 is a severe autosomal recessive form of the disorder.
Basement membrane-associated chondroitin sulfate proteoglycan (CSPG). Has prolyl 3-hydroxylase activity catalyzing the post-translational formation of 3-hydroxyproline in -Xaa-Pro-Gly- sequences in collagens, especially types IV and V. May be involved in the secretory pathway of cells. Has growth suppressive activity in fibroblasts
Endoplasmic reticulumSecreted, extracellular space, extracellular matrix
Osteogenesis imperfecta 8
A form of osteogenesis imperfecta, a disorder of bone formation characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI8 is characterized by disproportionate short stature, shortening of the long bones, white sclerae, a round face and a short barrel-shaped chest.
Chaperone specifically assisting the folding of beta-propeller/EGF modules within the family of low-density lipoprotein receptors (LDLRs) (PubMed:15014448). Acts as a modulator of the Wnt pathway through chaperoning the coreceptors of the canonical Wnt pathway, LRP5 and LRP6, to the plasma membrane (PubMed:17488095, PubMed:23572575). Essential for specification of embryonic polarity and mesoderm induction. Plays an essential role in neuromuscular junction (NMJ) formation by promoting cell-surfac
Endoplasmic reticulum
Osteogenesis imperfecta 20
An autosomal recessive form of osteogenesis imperfecta, a disorder of bone formation characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI20 is a progressive deforming form characterized by osteopenia, skeletal deformity, healed fractures, and newly-acquired fractures. Death due to respiratory failure can occur in some patients.
Necessary for efficient 3-hydroxylation of fibrillar collagen prolyl residues
Secreted, extracellular space, extracellular matrix
Osteogenesis imperfecta 7
A form of osteogenesis imperfecta, a disorder of bone formation characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI7 is an autosomal recessive, severe form. Multiple fractures are present at birth and patients have short stature, short humeri and femora, coxa vara, and white sclera. Dentinogenesis imperfecta is absent. Death can occur in the perinatal period due to secondary respiratory insufficiency.
Variantes genéticas (ClinVar)
337 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 7.473 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
29 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Osteogênese imperfeita tipo 2
Centros de Referência SUS
24 centros habilitados pelo SUS para Osteogênese imperfeita tipo 2
Centros para Osteogênese imperfeita tipo 2
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
Pesquisa e ensaios clínicos
1 ensaios clínicos encontrados.
Publicações mais relevantes
Fatality owing to pulmonary hemorrhage following pamidronate disodium administration in a neonate with osteogenesis imperfecta type 2: A case report.
We report the case of a patient with osteogenesis imperfecta (OI) who developed pulmonary hemorrhage 4 d after pamidronate disodium (PA) administration, despite a relatively stable respiratory status. Bisphosphonates are introduced to reduce osteoclast activity and are now widely used in patients with OI. Bisphosphonates are typically well-tolerated in children, and the standard of care involves cyclic intravenous administration of PA. However, in practice, there is limited experience with the use of PA for severe OI during the neonatal period, and its safety remains uncertain. This report aimed to describe the respiratory events potentially associated with PA in a neonatal patient with OI type 2, suggesting that serious life-threatening complications of pulmonary hemorrhage may occur after PA administration. Further studies are required to assess the relationship between pulmonary hemorrhage and PA administration, aiming to enhance prophylaxis measures.
A prospective study on rapid exome sequencing as a diagnostic test for multiple congenital anomalies on fetal ultrasound.
Conventional genetic tests (quantitative fluorescent-PCR [QF-PCR] and single nucleotide polymorphism-array) only diagnose ~40% of fetuses showing ultrasound abnormalities. Rapid exome sequencing (rES) may improve this diagnostic yield, but includes challenges such as uncertainties in fetal phenotyping, variant interpretation, incidental unsolicited findings, and rapid turnaround times. In this study, we implemented rES in prenatal care to increase diagnostic yield. We prospectively studied 55 fetuses. Inclusion criteria were: (a) two or more independent major fetal anomalies, (b) hydrops fetalis or bilateral renal cysts alone, or (c) one major fetal anomaly and a first-degree relative with the same anomaly. In addition to conventional genetic tests, we performed trio rES analysis using a custom virtual gene panel of ~3850 Online Mendelian Inheritance in Man (OMIM) genes. We established a genetic rES-based diagnosis in 8 out of 23 fetuses (35%) without QF-PCR or array abnormalities. Diagnoses included MIRAGE (SAMD9), Zellweger (PEX1), Walker-Warburg (POMGNT1), Noonan (PTNP11), Kabuki (KMT2D), and CHARGE (CHD7) syndrome and two cases of Osteogenesis Imperfecta type 2 (COL1A1). In six cases, rES diagnosis aided perinatal management. The median turnaround time was 14 (range 8-20) days. Implementing rES as a routine test in the prenatal setting is challenging but technically feasible, with a promising diagnostic yield and significant clinical relevance.
Comparative X-ray morphometry of prenatal osteogenesis imperfecta type 2 and thanatophoric dysplasia: a contribution to prenatal differential diagnosis.
The purpose of the paper was to assess the morphometric parameters to improve the specificity of the ultrasound (US) signs for the early differential diagnosis between two lethal dysplasias, as thanatophoric dysplasia (TD) and osteogenesis imperfecta type 2 (OI-2). The diaphyseal length and the bowed shape of long bones associated with vertebral body dimension assessment were investigated in a group of 14 pregnancy terminations carried out in the time period 2007-2013. The definitive diagnosis was established after pregnancy termination by means of skeletal standardized X-rays, histopathology and gene analysis. TD and OI-2 long bones were significantly shorter than controls. No significant differences were observed between the two dysplasias. The bowing angle was higher in OI-2; a true angulation or eventually axial displacement was present only in the latter. Furthermore, they did not show any evidence of vertebral collapse. The thanatophoric dysplasia presented less bowed long bones, and never true angulation. The spine was steadily characterized by flattened anterior vertebral bodies. Long bone shortening is not a sufficient and accurate sign for early sonographic differential diagnosis between TD and OI-2. Angled diaphysis, axial diaphyseal displacement and a conserved vertebral body height in the prenatal period support the diagnosis of osteogenesis imperfecta type 2, while moderately regular bowed diaphysis associated with platyspondyly that of thanatophoric dysplasia.
Publicações recentes
Fatality owing to pulmonary hemorrhage following pamidronate disodium administration in a neonate with osteogenesis imperfecta type 2: A case report.
A prospective study on rapid exome sequencing as a diagnostic test for multiple congenital anomalies on fetal ultrasound.
Comparative X-ray morphometry of prenatal osteogenesis imperfecta type 2 and thanatophoric dysplasia: a contribution to prenatal differential diagnosis.
Rare sonographic finding of osteogenesis imperfecta type 2: fluid retention in the subarachnoid space.
Genetic skeletal disorders of the fetus and infant: pathologic and molecular findings in a series of 41 cases.
📚 EuropePMC4.738 artigos no totalmostrando 3
Fatality owing to pulmonary hemorrhage following pamidronate disodium administration in a neonate with osteogenesis imperfecta type 2: A case report.
Clinical pediatric endocrinology : case reports and clinical investigations : official journal of the Japanese Society for Pediatric EndocrinologyA prospective study on rapid exome sequencing as a diagnostic test for multiple congenital anomalies on fetal ultrasound.
Prenatal diagnosisComparative X-ray morphometry of prenatal osteogenesis imperfecta type 2 and thanatophoric dysplasia: a contribution to prenatal differential diagnosis.
La Radiologia medicaAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Osteogênese imperfeita tipo 2.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Osteogênese imperfeita tipo 2
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Fatality owing to pulmonary hemorrhage following pamidronate disodium administration in a neonate with osteogenesis imperfecta type 2: A case report.Clinical pediatric endocrinology : case reports and clinical investigations : official journal of the Japanese Society for Pediatric Endocrinology· 2024· PMID 38572388mais citado
- A prospective study on rapid exome sequencing as a diagnostic test for multiple congenital anomalies on fetal ultrasound.
- Comparative X-ray morphometry of prenatal osteogenesis imperfecta type 2 and thanatophoric dysplasia: a contribution to prenatal differential diagnosis.
- Rare sonographic finding of osteogenesis imperfecta type 2: fluid retention in the subarachnoid space.
- Genetic skeletal disorders of the fetus and infant: pathologic and molecular findings in a series of 41 cases.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:216804(Orphanet)
- OMIM OMIM:166210(OMIM)
- MONDO:0008147(MONDO)
- GARD:10142(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q27677736(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
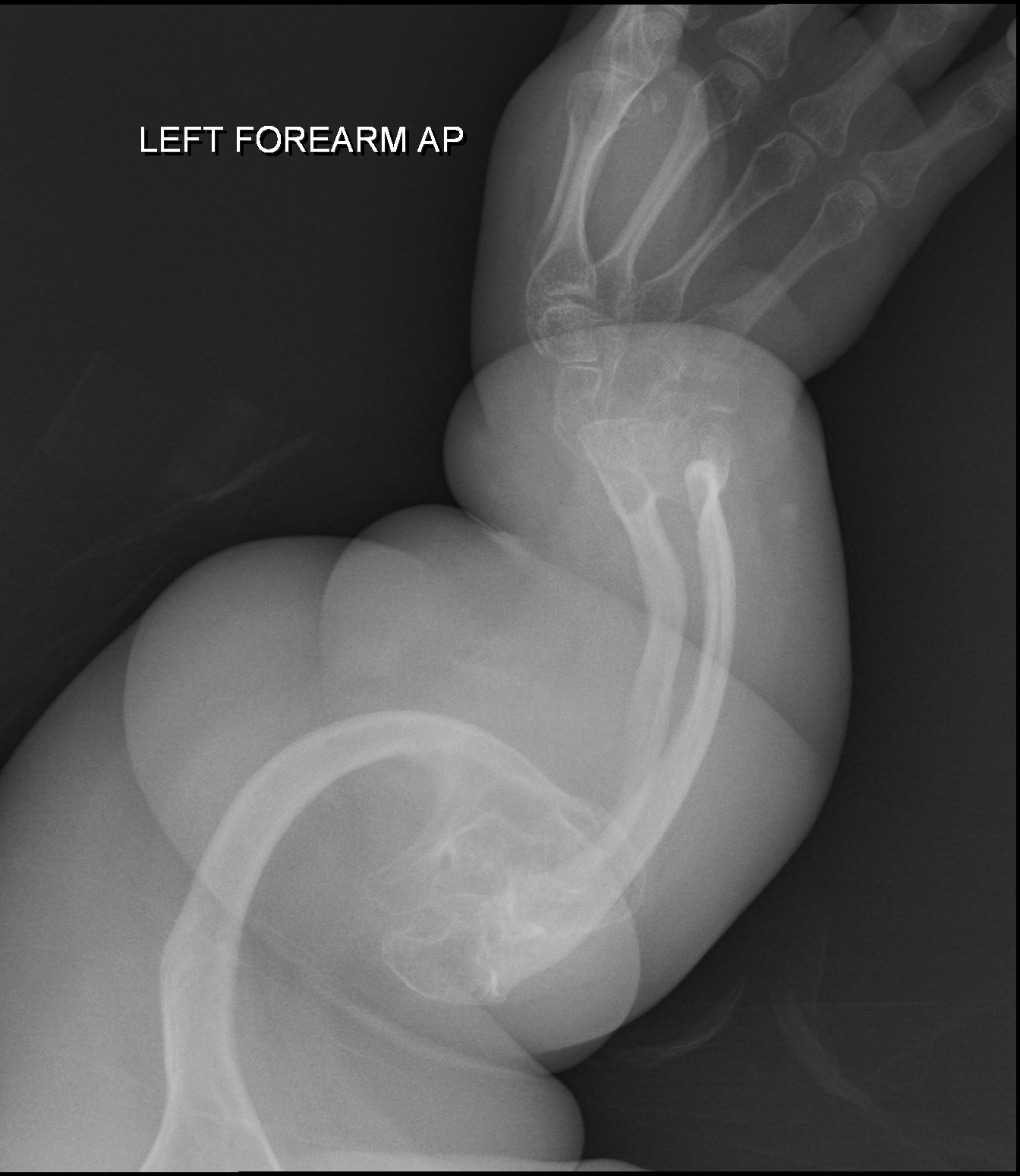
Osteogênese imperfeita tipo 2
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata