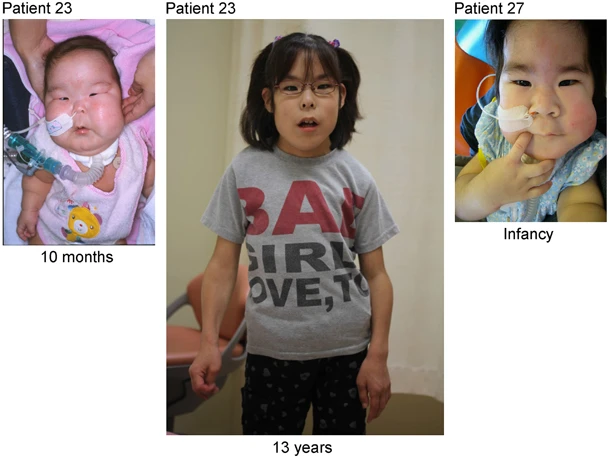
Doença genética rara caracterizada por polidrâmnio (principalmente devido à placentomegalia), macrossomia fetal, defeitos da parede abdominal, anormalidades esqueléticas (incluindo tórax em forma de sino, aparência de cabide das costelas e diminuição da proporção do diâmetro médio a largo do tórax na infância), dificuldades de alimentação e dificuldade de deglutição, características dismórficas (testa cabeluda, bochechas cheias, filtro protuberante, micrognatia), atraso no desenvolvimento e deficiência intelectual. Características adicionais podem incluir cifoescoliose, contraturas articulares, diástase retal e hipotonia muscular. Existe risco aumentado de hepatoblastoma.
Introdução
O que você precisa saber de cara
Doença genética rara caracterizada por polidrâmnio (principalmente devido à placentomegalia), macrossomia fetal, defeitos da parede abdominal, anormalidades esqueléticas (incluindo tórax em forma de sino, aparência de cabide das costelas e diminuição da proporção do diâmetro médio a largo do tórax na infância), dificuldades de alimentação e dificuldade de deglutição, características dismórficas (testa cabeluda, bochechas cheias, filtro protuberante, micrognatia), atraso no desenvolvimento e deficiência intelectual. Características adicionais podem incluir cifoescoliose, contraturas articulares, diástase retal e hipotonia muscular. Existe risco aumentado de hepatoblastoma.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 61 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 158 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
3 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Not applicable.
May have a role in neuroendocrine differentiation
MembraneCytoplasm
Plays an essential role in capillaries endothelial cells for the maintenance of feto-maternal interface and for development of the placenta
Membrane
Variantes genéticas (ClinVar)
129 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Anomalias congênitas múltiplas por anomalia nos genes de expressão materna localizados em 14q32.2
Centros de Referência SUS
24 centros habilitados pelo SUS para Anomalias congênitas múltiplas por anomalia nos genes de expressão materna localizados em 14q32.2
Centros para Anomalias congênitas múltiplas por anomalia nos genes de expressão materna localizados em 14q32.2
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
Prenatal suspicion of Kagami-Ogata syndrome based on Robertsonian translocation (14;22).
Kagami-Ogata syndrome is characterized by developmental delay, intellectual disability, feeding difficulty with impaired swallowing, full cheeks, prominent and deep philtrum, small bell-shaped thorax with coat-hanger appearance of the ribs, and abdominal wall defects (omphalocele and diastasis recti). Additional common features include joint contractures, kyphoscoliosis, coxa valga, and laryngomalacia. Cardiac disease and hepatoblastoma have also been reported. The diagnosis of Kagami-Ogata syndrome is established in a proband with suggestive findings and findings on molecular genetic testing that suggest deficient expression of the RTL1 antisense (RTL1as) allele of maternal origin due to one of the following: paternal uniparental disomy of chromosome 14; epimutation (hypermethylation) affecting the normally unmethylated MEG3/DLK1 intergenic differentially methylated region (MEG3/DLK1:IG-DMR) and MEG3 transcription start site DMR (MEG3:TSS-DMR) on the maternal allele; deletion of the maternally inherited 14q32.2 region that includes MEG3/DLK1:IG-DMR and/or MEG3:TSS-DMR, and may include RTL1as; deletion of the maternally inherited RTL1as but not MEG3/DLK1:IG-DMR or MEG3:TSS-DMR; translocation (or inversion) disrupting the integrity between the maternally inherited MEG3 promoter at the MEG3:TSS-DMR and RTL1as. Treatment of manifestations: Developmental and educational support; mechanical ventilation and oxygen therapy as needed after birth; tracheostomy as required; monitor and treat upper and lower respiratory tract infections; treatment of scoliosis per orthopedist; treatment of joint contractures per rehabilitation medicine specialist; tube feeding typically required for poor weight gain; gastrostomy tube feeding as needed; feeding training; treatment of cardiac disease per cardiologist; standard treatment of hepatoblastoma with surgery and chemotherapy; social work and family support. Surveillance: Monitor developmental progress, educational needs, for evidence of aspiration or respiratory insufficiency, progression of kyphoscoliosis and joint contractures, nutritional status, safety of oral intake, constipation, and family and care coordination needs at each visit. Echocardiogram annually; abdominal ultrasound and serum alpha-fetoprotein every three months until age three to four years. Agents/circumstances to avoid: Avoid exposure to respiratory infections; individuals with Kagami-Ogata syndrome may develop respiratory failure with respiratory infections, especially during infancy. The recurrence risk of Kagami-Ogata syndrome is dependent on the genetic mechanism underlying deficient expression of the maternal RTL1as allele in the proband. In most affected individuals, the underlying genetic mechanism occurs as a de novo event and the recurrence risk to sibs is not increased. Less commonly, a parent of a proband has a predisposing genetic alteration associated with an increased recurrence risk to sibs. Recurrence of Kagami-Ogata syndrome has been reported in sibs with maternally derived deletions. If a deletion or translocation involving the chromosome 14q32.2 imprinted region has been identified in the proband, prenatal testing and preimplantation genetic testing are possible. Methylation testing of fetal DNA to examine abnormal methylation patterns of the DMRs (MEG3/DLK1:IG-DMR and MEG3:TSS-DMR) is not recommended; while DNA extracted from amniotic fluid is currently believed to provide the most reliable tissue source for evaluating fetal methylation status, false negative findings have been reported.
Comprehensive molecular and clinical findings in 29 patients with multi-locus imprinting disturbance.
Multi-locus imprinting disturbance (MLID) with methylation defects in various differentially methylated regions (DMRs) has recently been identified in approximately 150 cases with imprinting disorders (IDs), and deleterious variants have been found in genes related to methylation maintenance of DMRs, such as those encoding proteins constructing the subcortical maternal complex (SCMC), in a small fraction of patients and/or their mothers. However, integrated methylation analysis for DMRs and sequence analysis for MLID-causative genes in MLID cases and their mothers have been performed only in a single study focusing on Beckwith-Wiedemann syndrome (BWS) and Silver-Russell syndrome (SRS) phenotypes. Of 783 patients with various IDs we have identified to date, we examined a total of 386 patients with confirmed epimutation and 71 patients with epimutation or uniparental disomy. Consequently, we identified MLID in 29 patients with epimutation confirmed by methylation analysis for multiple ID-associated DMRs using pyrosequencing and/or methylation-specific multiple ligation-dependent probe amplification. MLID was detected in approximately 12% of patients with BWS phenotype and approximately 5% of patients with SRS phenotype, but not in patients with Kagami-Ogata syndrome, Prader-Willi syndrome, or Angelman syndrome phenotypes. We next conducted array-based methylation analysis for 78 DMRs and whole-exome sequencing in the 29 patients, revealing hypomethylation-dominant aberrant methylation patterns in various DMRs of all the patients, eight probably deleterious variants in genes for SCMC in the mothers of patients, and one homozygous deleterious variant in ZNF445 in one patient. These variants did not show gene-specific methylation disturbance patterns. Clinically, neurodevelopmental delay and/or intellectual developmental disorder (ND/IDD) was observed in about half of the MLID patients, with no association with the identified methylation disturbance patterns and genetic variants. Notably, seven patients with BWS phenotype were conceived by assisted reproductive technology (ART). The frequency of MLID was 7.5% (29/386) in IDs caused by confirmed epimutation. Furthermore, we revealed diverse patterns of hypomethylation-dominant methylation defects, nine deleterious variants, ND/IDD complications in about half of the MLID patients, and a high frequency of MLID in ART-conceived patients.
Prenatal diagnosis of recurrent Kagami-Ogata syndrome inherited from a mother affected by Temple syndrome: a case report and literature review.
Kagami-Ogata syndrome (KOS) and Temple syndrome (TS) are two imprinting disorders characterized by the absence or reduced expression of maternal or paternal genes in the chromosome 14q32 region, respectively. We present a rare prenatally diagnosed case of recurrent KOS inherited from a mother affected by TS. The woman's two affected pregnancies exhibited recurrent manifestations of prenatal overgrowth, polyhydramnios, and omphalocele, as well as a small bell-shaped thorax with coat-hanger ribs postnatally. Prenatal genetic testing using a single-nucleotide polymorphism array detected a 268.2-kb deletion in the chromosome 14q32 imprinted region inherited from the mother, leading to the diagnosis of KOS. Additionally, the woman carried a de novo deletion in the paternal chromosome 14q32 imprinted region and presented with short stature and small hands and feet, indicating a diagnosis of TS. Given the rarity of KOS as an imprinting disorder, accurate prenatal diagnosis of this rare imprinting disorder depends on two factors: (1) increasing clinician recognition of the clinical phenotype and related genetic mechanism, and (2) emphasizing the importance of imprinted regions in the CMA workflow for laboratory analysis.
Adults with paternal UPD14 causing Kagami-Ogata syndrome: Case report and review of the literature.
Kagami-Ogata syndrome (KOS) is a clinically recognizable syndrome in the neonatal period. It is characterized by specific skeletal anomalies and facial dysmorphisms. It is typically caused by paternal uniparental disomy of chromosome 14, while epimutations and microdeletions are less commonly reported causes. In the pediatric setting, KOS is a well delineated syndrome. However, there is a dearth of literature describing the natural history of the condition in adults. Herein, we describe a 35-year-old man, the first adult with KOS reported due to paternal uniparental disomy 14, and review reports of KOS in other affected adults. This highlights the variability in neurocognitive phenotypes, the presence of connective tissue abnormalities, and the uncertainties around long-term cancer risk.
Polyhydramnios associated with rare genetic syndromes: two case reports.
We present two genetic causes of polyhydramnios that were challenging to diagnose due to their rarity and complexity. In view of the severe implications, we wish to highlight these rare genetic conditions when obstetricians consider differential diagnoses of polyhydramnios in the third trimester. Patient 1 is a 34-year-old Asian woman who was diagnosed with polyhydramnios at 28 weeks' gestation. First trimester testing, fetal anomaly scan, and intrauterine infection screen were normal. Subsequent antenatal ultrasound scans revealed macroglossia, raising the suspicion for Beckwith-Wiedemann syndrome. Chromosomal microarray analysis revealed a female profile with no pathological copy number variants. The patient underwent amnioreduction twice in the pregnancy. The patient presented in preterm labor at 34 weeks' gestation but elected for an emergency caesarean section. Postnatally, the baby was noted to have a bell-shaped thorax, coat hanger ribs, hypotonia, abdominal distension, and facial dysmorphisms suggestive of Kagami-Ogata syndrome. Patient 2 is a 30-year-old Asian woman who was diagnosed with polyhydramnios at 30 weeks' gestation. She had a high-risk first trimester screen but declined invasive testing; non-invasive prenatal testing was low risk. Ultrasound examination revealed a macrosomic fetus with grade 1 echogenic bowels but no other abnormalities. Intrauterine infection screen was negative, and there was no sonographic evidence of fetal anemia. She had spontaneous rupture of membranes at 37 + 3 weeks but subsequently delivered by caesarean section in view of pathological cardiotocography. The baby was noted to have inspiratory stridor, hypotonia, low-set ears, and bilateral toe polysyndactyly. Further genetic testing revealed a female profile with a pathogenic variant of the GLI3 gene, confirming a diagnosis of Greig cephalopolysyndactyly syndrome. These cases illustrate the importance of considering rare genetic causes of polyhydramnios in the differential diagnosis, particularly when fetal anomalies are not apparent at the 20-week structural scan. We would like to raise awareness for these rare conditions, as a high index of suspicion enables appropriate counseling, prenatal testing, and timely referral to pediatricians and geneticists. Early identification and diagnosis allow planning of perinatal care and birth in a tertiary center managed by a multidisciplinary team.
Publicações recentes
Prenatal suspicion of Kagami-Ogata syndrome based on Robertsonian translocation (14;22).
Comprehensive molecular and clinical findings in 29 patients with multi-locus imprinting disturbance.
Prenatal diagnosis of recurrent Kagami-Ogata syndrome inherited from a mother affected by Temple syndrome: a case report and literature review.
Adults with paternal UPD14 causing Kagami-Ogata syndrome: Case report and review of the literature.
📚 EuropePMC33 artigos no totalmostrando 42
Prenatal suspicion of Kagami-Ogata syndrome based on Robertsonian translocation (14;22).
Pediatrics international : official journal of the Japan Pediatric SocietyComprehensive molecular and clinical findings in 29 patients with multi-locus imprinting disturbance.
Clinical epigeneticsPrenatal diagnosis of recurrent Kagami-Ogata syndrome inherited from a mother affected by Temple syndrome: a case report and literature review.
BMC medical genomicsAdults with paternal UPD14 causing Kagami-Ogata syndrome: Case report and review of the literature.
American journal of medical genetics. Part APolyhydramnios associated with rare genetic syndromes: two case reports.
Journal of medical case reportsKagami Ogata syndrome: a small deletion refines critical region for imprinting.
NPJ genomic medicineImprinted small nucleolar RNAs: Missing link in development and disease?
Wiley interdisciplinary reviews. RNAMaternally inherited deletion encompassing the RTL1as and MEG8 genes of the human 14q32 imprinted region in a patient with a mild Kagami-Ogata syndrome phenotype.
American journal of medical genetics. Part APaternal UPD14 with sSMC derived from chromosome 14 in Kagami-Ogata syndrome.
Chromosome research : an international journal on the molecular, supramolecular and evolutionary aspects of chromosome biologyQuantitative assessment of coat-hanger ribs detected on three-dimensional ultrasound for prenatal diagnosis of Kagami-Ogata syndrome.
The journal of obstetrics and gynaecology researchCase report: Prenatal diagnosis of Kagami-Ogata syndrome in a Chinese family.
Frontiers in geneticsTwo infants with mild, atypical clinical features of Kagami-Ogata syndrome caused by epimutation.
European journal of medical geneticsKagami-Ogata syndrome: a case report.
Journal of medical case reportsFirst prenatal case of Kagami-Ogata syndrome associated with a small supernumerary marker chromosome derived from chromosome 15.
Taiwanese journal of obstetrics & gynecologyPrenatal Diagnosis of a Mosaic Paternal Uniparental Disomy for Chromosome 14: A Case Report of Kagami-Ogata Syndrome.
Frontiers in pediatricsKagami-Ogata Syndrome: Case Series and Review of Literature.
AJP reportsPreimplantation genetic testing for a chr14q32 microdeletion in a family with Kagami-Ogata syndrome and Temple syndrome.
Journal of medical geneticsAnesthetic management of a 12-year-old child with Kagami-Ogata syndrome for pectus excavatum: a case report.
JA clinical reportsPrenatal diagnosis of Kagami-Ogata syndrome.
Journal of clinical ultrasound : JCUKagami-Ogata syndrome in a patient with 46,XX,t(2;14)(q11.2;q32.2)mat disrupting MEG3.
Journal of human geneticsDeficiency and overexpression of Rtl1 in the mouse cause distinct muscle abnormalities related to Temple and Kagami-Ogata syndromes.
Development (Cambridge, England)Temple syndrome and Kagami-Ogata syndrome: clinical presentations, genotypes, models and mechanisms.
Human molecular geneticsA Male Case of Kagami-Ogata Syndrome Caused by Paternal Unipaternal Disomy 14 as a Result of a Robertsonian Translocation.
Frontiers in pediatricsKagami-Ogata syndrome: an important differential diagnosis to Beckwith-Wiedemann syndrome.
Journal of clinical ultrasound : JCU[Kagami-Ogata Syndrome: An Anomaly of the Ribs as a Pathognomonic Feature for the Clinical Diagnosis of an (epi)Genetic Syndrome].
Zeitschrift fur Geburtshilfe und NeonatologieKagami-Ogata syndrome in a fetus presenting with polyhydramnios, malformations, and preterm delivery: a case report.
Journal of medical case reportsSingle-nucleotide polymorphism-based chromosomal microarray analysis provides clues and insights into disease mechanisms.
Ultrasound in obstetrics & gynecology : the official journal of the International Society of Ultrasound in Obstetrics and GynecologyMaternally inherited 133kb deletion of 14q32 causing Kagami-Ogata syndrome.
Journal of human geneticsAnesthetic management of a child with Kagami-Ogata syndrome complicated with marked tracheal deviation: a case report.
JA clinical reportsSnord116-dependent diurnal rhythm of DNA methylation in mouse cortex.
Nature communicationsMosaic upd(14)pat in a patient with mild features of Kagami-Ogata syndrome.
Clinical case reportsDLK1-DIO3 imprinted locus deregulation in development, respiratory disease, and cancer.
Expert review of respiratory medicineTemple syndrome: comprehensive molecular and clinical findings in 32 Japanese patients.
Genetics in medicine : official journal of the American College of Medical GeneticsNew insights into the imprinted MEG8-DMR in 14q32 and clinical and molecular description of novel patients with Temple syndrome.
European journal of human genetics : EJHGFamilial Kagami-Ogata syndrome in Chinese.
Clinical dysmorphologyGenome-wide multilocus imprinting disturbance analysis in Temple syndrome and Kagami-Ogata syndrome.
Genetics in medicine : official journal of the American College of Medical GeneticsChest Radiograph as Diagnostic Clue in a Floppy Infant.
The Journal of pediatricsNovel microdeletions on chromosome 14q32.2 suggest a potential role for non-coding RNAs in Kagami-Ogata syndrome.
European journal of human genetics : EJHGThe Coat-Hanger Angle Sign.
The Journal of pediatricsKagami-Ogata syndrome: a clinically recognizable upd(14)pat and related disorder affecting the chromosome 14q32.2 imprinted region.
Journal of human geneticsExploration of hydroxymethylation in Kagami-Ogata syndrome caused by hypermethylation of imprinting control regions.
Clinical epigeneticsComprehensive clinical studies in 34 patients with molecularly defined UPD(14)pat and related conditions (Kagami-Ogata syndrome).
European journal of human genetics : EJHGAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Anomalias congênitas múltiplas por anomalia nos genes de expressão materna localizados em 14q32.2.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Anomalias congênitas múltiplas por anomalia nos genes de expressão materna localizados em 14q32.2
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Prenatal suspicion of Kagami-Ogata syndrome based on Robertsonian translocation (14;22).Pediatrics international : official journal of the Japan Pediatric Society· 2025· PMID 41246872mais citado
- Comprehensive molecular and clinical findings in 29 patients with multi-locus imprinting disturbance.
- Prenatal diagnosis of recurrent Kagami-Ogata syndrome inherited from a mother affected by Temple syndrome: a case report and literature review.
- Adults with paternal UPD14 causing Kagami-Ogata syndrome: Case report and review of the literature.
- Polyhydramnios associated with rare genetic syndromes: two case reports.
- Kagami-Ogata Syndrome.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:254519(Orphanet)
- MONDO:0016779(MONDO)
- GARD:17219(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q56013887(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Anomalias congênitas múltiplas por anomalia nos genes de expressão materna localizados em 14q32.2
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov