A Síndrome 47, XYY é uma condição genética que acontece por uma alteração nos cromossomos sexuais, onde homens recebem um cromossomo Y extra. Ela é caracterizada por altura acima da média, notável desde a infância, cabeça maior que o normal e características faciais específicas, como olhos um pouco mais afastados, orelhas com implantação um pouco mais baixa e maçãs do rosto levemente mais planas. Além disso, pode haver atraso na fala e um risco maior de desenvolver dificuldades sociais e emocionais, Transtorno de Déficit de Atenção e Hiperatividade (TDAH) e Transtorno do Espectro Autista (TEA).
Introdução
O que você precisa saber de cara
A Síndrome 47, XYY é uma condição genética que acontece por uma alteração nos cromossomos sexuais, onde homens recebem um cromossomo Y extra. Ela é caracterizada por altura acima da média, notável desde a infância, cabeça maior que o normal e características faciais específicas, como olhos um pouco mais afastados, orelhas com implantação um pouco mais baixa e maçãs do rosto levemente mais planas. Além disso, pode haver atraso na fala e um risco maior de desenvolver dificuldades sociais e emocionais, Transtorno de Déficit de Atenção e Hiperatividade (TDAH) e Transtorno do Espectro Autista (TEA).
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 19 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 45 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome 47,XYY
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
32 ensaios clínicos encontrados, 1 ativos.
Publicações mais relevantes
Neuropsychiatric Phenotype and Treatment Challenges in 47,XYY Syndrome: A Narrative Review with a Case Series of Adolescents.
47,XYY syndrome is a relatively common sex chromosome aneuploidy that remains largely underdiagnosed. While its somatic phenotype is often mild, growing evidence indicates a substantial burden of neurodevelopmental and psychiatric morbidity. However, the characterization of the neuropsychiatric phenotype across development, particularly during adolescence, and the associated treatment challenges remain incomplete. To provide a comprehensive narrative review of the neuropsychiatric phenotype of 47,XYY syndrome and to illustrate clinical complexity and treatment response through a case series of adolescents. A narrative review of the literature was conducted focusing on genetics, neurodevelopmental and psychiatric features, neuroimaging and neurophysiology findings, clinical course, and management strategies in 47,XYY syndrome. This review is complemented by a case series of adolescents with confirmed 47,XYY karyotype, evaluated for developmental history, psychiatric comorbidity and response to pharmacological and non-pharmacological interventions. The literature consistently describes increased risks of language impairment, executive dysfunction, ADHD, autism spectrum traits, and emotional and behavioral dysregulation in males with 47,XYY syndrome. Psychiatric vulnerability appears to increase during adolescence and adulthood, with elevated rates of mood, psychotic, and substance use disorders. The presented cases illustrate a convergent clinical trajectory marked by early developmental delays, progressive behavioral dysregulation in adolescence and limited or inconsistent response to multiple classes of psychotropic medications, suggesting a pattern of pharmacoresistance in a subset of patients. 47,XYY syndrome is associated with a distinct and heterogeneous neuropsychiatric phenotype that extends beyond early neurodevelopmental disorders. Early diagnosis alone may be insufficient to prevent severe psychiatric outcomes, highlighting the need for long-term monitoring and integrated, multidisciplinary management. Further research is required to identify early predictors of high-risk trajectories and to optimize treatment strategies for this population.
Neurodevelopmental and Mental Health Outcomes in a National Clinical Sample of Youth With Sex Chromosome Trisomies Compared With Matched Controls.
To compare the prevalence of neurodevelopmental and mental health diagnoses in a national sample of youth with sex chromosome trisomies (SCTs) with matched controls. Patients in PEDSnet and a diagnosis code mapping to 47,XXY/Klinefelter syndrome (n = 1171), 47,XYY/Double Y syndrome (n = 243), or 47,XXX/Trisomy X syndrome (n = 262) were matched with controls using propensity scores. Generalized estimating equations computed odds ratios (OR) with 95% confidence intervals (CI) for the prevalence of diagnoses within the neurodevelopmental and mental health composites, psychotropic medication prescriptions, and encounters with behavioral health and therapy providers. Alpha was set at 0.0025 to account for multiple comparisons. Patients with SCTs had higher odds of diagnoses within the neurodevelopmental (OR 6.3, 95% CI, 5.7-7.2) and mental health composites (OR 2.7, 95% CI, 2.3-3.2) compared with matched controls. All neurodevelopmental diagnoses were more prevalent among all SCT groups compared with controls. Within the mental health composite, only the prevalence of anxiety and mood disorder was higher in all SCT groups. A higher proportion of patients with SCTs had psychotropic prescriptions compared with controls (stimulants 13.1% vs 5.2%, selective serotonin reuptake inhibitors 8.7% vs 2.8%, antipsychotics 6.5% vs 2.4%, p < 0.0001 for all). Overall, 48% of patients with SCTs had a clinical encounter with a behavioral health provider vs 16.6% of controls (OR 5.6, 95% CI, 4.1-5.1). Compared with matched controls, youth with SCTs receiving care at US tertiary care pediatric centers have disproportionately high rates of neurodevelopmental and mental health conditions, emphasizing the need for appropriate screening and intervention in these populations.
Can Individuals with 47,XYY Karyotypes Exist without Male Phenotype? A Narrative Literature Review and Case Report.
The 47,XYY syndrome is a genetic condition found in about 1 in 1000 male children. The expected phenotype is male but could vary greatly. Those with genitourinary abnormalities may also present with microphallus, hypoplastic scrotum, cryptorchidism, hypospadias and macroorchidism. This study reports a child with sex ambiguity who possesses an initial 47,XYY karyotype. We also conducted a narrative literature review of 47,XYY individuals and their respective genital phenotype and/or gender identity. The narrative literature review was performed by searching for "47,XYY" in the PubMed database. All studies published in English, Spanish or Portuguese from January 1960 to January 2024 that contained the term "47,XYY" in the title or abstract were included. Studies that did not describe the genital phenotype and/or gender identity of cases were excluded. We also described the case of a 2-month-old patient with the 47,XYY karyotype and sex ambiguity. Our patient underwent additional karyotype testing, resulting in 47,XYY [30] and another 45,X [2]/47,XYY [98] with mosaicism being confirmed by fluorescent in situ hybridization (FISH) on buccal smears (nuc ish (DXZ1 × 1, DYZ3 × 2)[64/100]/(DXZ1 × 1, DYZ3 × 0)[36/100]. A gonadal biopsy revealed an atrophic testis on the left and a streak gonad on the right, with a final diagnosis of mixed gonadal dysgenesis determined. The narrative review revealed 643 articles, of which 350 met the inclusion criteria. However, we excluded 132 articles because they presented no new cases. We included 138 articles, which presented a series containing less than 10 new cases with the 47,XYY karyotype (total of 327 cases), 58 articles presented 4001 cases and 22 articles presented 75 patients with the 47,XYY karyotype in mosaic with 45,X. For all 4403 analyzed cases, 4354 (98.90%) presented a male phenotype, of which 4322 had the 47,XYY karyotype and 32 had mosaicism with 45,X lineage. A further 23 (0.52%) presented a female phenotype, of which four had the 47,XYY karyotype and 19 had mosaicism with 45,X lineage. In addition, 23 (0.52%) cases presented ambiguous genitalia, of which two had the 47,XYY karyotype and 21 had mosaicism with 45,X lineage. Finally, three (0.06%) cases had undefined phenotypes, all with mosaicism with 45,X lineage. Of the six cases with the 47,XYY karyotype and no male phenotype, one had complete androgen insensitivity syndrome (CAIS), one had lipoid congenital adrenal hyperplasia, two had probable CAIS, and two presented an incomplete diagnostic investigation. A female or ambiguous genital phenotype in an individual with 47,XYY karyotype is uncommon and should alert to the presence of the 45,X lineage or association with other causes of disorder/difference of sex development.
[Clinical characteristics and genetic analysis of a case with 47,XYY Disorder of sex development due to variant of NR5A1 gene].
To investigate the clinical phenotype and genetic etiology of a patient with tall stature and primary amenorrhea presenting with 47,XYY Disorder of sex development (DSD). A female patient presenting with "tall stature and primary amenorrhea" at Nanjing Drum Tower Hospital in July 2024 was selected as the study subject. A retrospective study design was employed to collect the patient's clinical data. Peripheral venous blood sample was collected. Following the extraction of genomic DNA, genetic testing was performed including chromosomal karyotyping analysis, copy number variation sequencing (CNV-seq), multiplex PCR for the AZF regions and sex-determining genes Y (SRY), and whole-exome sequencing (WES). Candidate variants were validated by Sanger sequencing and classified for pathogenicity based on the guidelines from the American College of Medical Genetics and Genomics (ACMG). This study was approved by the Medical Ethics Committee of Nanjing Drum Tower Hospital (Ethics No.: 2022-451-01). The patient had a height of 188 cm and a body weight of 50 kg, in addition with infantile uterus, absent ovaries, and primary amenorrhea. G-banded karyotyping analysis of peripheral blood sample revealed 47,XYY. CNV-seq indicated Seq[GRCh37]Yp11.32q12×2. No deletion was detected in the AZF regions of Y chromosome, and SRY was positive. WES identified a heterozygous c.86C>A (p.Thr29Lys) variant of the NR5A1 gene, leading to substitution of threonine with lysine at position 29 of the encoded protein. Sanger sequencing confirmed the presence of the variant. According to the ACMG guidelines, this variant was classified as variant of uncertain significance (VUS) with supporting evidence (PS3_Moderate+PM5+PP3+PM2_Supporting+PS4_Supporting). Reviewing the nearly 60 years of previously reported cases, all 7 documented 47,XYY DSD patients were assigned a female social gender and presented with abnormal gonadal and external genitalia development. Among them, 5 cases underwent SRY testing, all of which were positive. Only 1 case underwent whole-exome sequencing (WES), but no pathogenic or likely pathogenic variants were identified. This DSD patient presented with the clinical features of tall stature and primary amenorrhea. The NR5A1 gene variant c.86C>A (p.Thr29Lys) probably underlay the Disorder of sex development in this patient. Above finding has enriched the spectrum of pathogenic variants of the NR5A1 gene.
An iPSC-based model of 47,XYY Jacobs syndrome reveals a DNA methylation-independent transcriptional dysregulation shared with male X aneuploid cells.
Jacobs (JS) and Klinefelter (KS) syndromes, carrying 47,XYY and 47,XXY chromosomes, respectively, are the most prevalent sex-chromosome aneuploidies in males. JS and KS patients share several clinical features, including sterility, hormonal deficits, neurocognitive delay, and skeletal-muscle defects, although the penetrance of these traits in the two syndromes varies. Despite the high incidence, the molecular mechanisms underlying the clinical manifestations in sex aneuploid male patients are still elusive. In this study, we characterize the inaugural cohort of 47,XYY human induced pluripotent stem cells (iPSCs). We perform a comprehensive transcriptional analysis, including 47,XYY and 46,XY primary fibroblasts, iPSCs, and neural stem cells (NSCs), alongside a comparative analysis of 47,XYY and 47,XXY fibroblasts and iPSC transcriptomes. We reveal a transcriptional feedback mechanism tuning non-PAR X Chromosome gene (NPX) homologs in Y supernumerary cells, a phenomenon not detected in X aneuploid male iPSCs. By ectopically modulating the expression of selected NPY genes, we demonstrate a transcriptional link between the UTY-KDM6A gene pair. Furthermore, our analyses identify a shared transcriptomic signature between JS and KS, discernible already at the iPSC stage, with a notable enrichment for processes related to neurological functions. This transcriptomic convergence underscores potential commonalities in the molecular pathways underpinning the pathophysiology of male sex-chromosome aneuploidies. Finally, through genome-wide DNA methylation profiling of JS iPSCs, we demonstrate that a supernumerary Y Chromosome only minimally impacts the methylation status of 47,XYY cells at the pluripotent stage. Our work reveals critical transcriptional feedback mechanisms and shared gene expression signatures in male sex-chromosome aneuploidies.
Publicações recentes
Neuropsychiatric Phenotype and Treatment Challenges in 47,XYY Syndrome: A Narrative Review with a Case Series of Adolescents.
Preimplantation genetic testing might not be the necessity for male patients with 47,XYY syndrome: A pilot study.
📖 RevisãoCan Individuals with 47,XYY Karyotypes Exist without Male Phenotype? A Narrative Literature Review and Case Report.
Cytogenetic analysis of 3488 patients with recurrent pregnancy loss: An experience of two decades from a tertiary care center in South India.
Gonadal function in patients with 47,XYY syndrome: a systematic review and meta-analysis.
📚 EuropePMC43 artigos no totalmostrando 66
Neuropsychiatric Phenotype and Treatment Challenges in 47,XYY Syndrome: A Narrative Review with a Case Series of Adolescents.
Brain sciences[Clinical characteristics and genetic analysis of a case with 47,XYY Disorder of sex development due to variant of NR5A1 gene].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsAn iPSC-based model of 47,XYY Jacobs syndrome reveals a DNA methylation-independent transcriptional dysregulation shared with male X aneuploid cells.
Genome researchPreimplantation genetic testing might not be the necessity for male patients with 47,XYY syndrome: A pilot study.
Reproductive medicine and biologyNeurodevelopmental and Mental Health Outcomes in a National Clinical Sample of Youth With Sex Chromosome Trisomies Compared With Matched Controls.
Journal of developmental and behavioral pediatrics : JDBPCan Individuals with 47,XYY Karyotypes Exist without Male Phenotype? A Narrative Literature Review and Case Report.
Frontiers in bioscience (Scholar edition)A new case with coexistence of mosaic 48,XYYY/47,XYY, and CACNA1E variant in autism spectrum disorder.
Psychiatric geneticsReaction to Diagnosis and Parental Concerns in Parents of Children and Young Adults With XYY Syndrome.
Child: care, health and developmentPrevalence, Morbidity, and Mortality of Men With Sex Chromosome Aneuploidy in the Million Veteran Program Cohort.
JAMA network openClinical evaluation of noninvasive prenatal testing for sex chromosome aneuploidies in 9,176 Korean pregnant women: a single-center retrospective study.
BMC pregnancy and childbirthDeep phenotypic analysis of psychiatric features in genetically defined cohorts: application to XYY syndrome.
Journal of neurodevelopmental disordersNew developments and future trajectories in supernumerary sex chromosome abnormalities: a summary of the 2022 3rd International Workshop on Klinefelter Syndrome, Trisomy X, and XYY.
Endocrine connectionsAssessing the value of second-trimester nasal bone hypoplasia in predicting chromosomal abnormalities: a retrospective chromosomal microarray analysis of 351 fetuses.
Archives of gynecology and obstetrics[Genetic analysis of a child with XYY syndrome in conjunct with 3-methylglutaenedioic aciduria type I].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsA patient with 47, XYY mosaic karyotype and congenital absence of bilateral vas deferens: a case report and literature review.
BMC urologyDNA methylation and behavioral dysfunction in males with 47,XXY and 49,XXXXY: a pilot study.
Clinical epigeneticsDecreased levels of γ-aminobutyric acid in temporal lobe of children with 47,XYY syndrome.
NeuroreportModeling familial predictors of proband outcomes in neurogenetic disorders: initial application in XYY syndrome.
Journal of neurodevelopmental disordersMEIS2 sequence variant in a child with intellectual disability and cardiac defects: Expansion of the phenotypic spectrum and documentation of low-level mosaicism in an unaffected parent.
American journal of medical genetics. Part A[Association between karyotype 47XYY and 5-alpha reductase deficiency revealed by micropenis: about a case and literature review].
The Pan African medical journalTesticular function in boys with 47,XYY and relationship to phenotype.
American journal of medical genetics. Part C, Seminars in medical geneticsThe epidemiology of sex chromosome abnormalities.
American journal of medical genetics. Part C, Seminars in medical geneticsEarly neurodevelopmental and medical profile in children with sex chromosome trisomies: Background for the prospective eXtraordinarY babies study to identify early risk factors and targets for intervention.
American journal of medical genetics. Part C, Seminars in medical geneticsUsefulness of methylation-specific multiplex ligation-dependent probe amplification for identification of parental origin of triploidy.
Journal of human geneticsMorbidity in 47,XYY syndrome: a nationwide epidemiological study of hospital diagnoses and medication use.
Genetics in medicine : official journal of the American College of Medical GeneticsThe behavioral profile of children aged 1-5 years with sex chromosome trisomy (47,XXX, 47,XXY, 47,XYY).
American journal of medical genetics. Part C, Seminars in medical geneticsMeiotic Behavior of Extra Sex Chromosomes in Patients with the 47,XXY and 47,XYY Karyotype and Its Ultimate Consequences for Spermatogenesis.
Critical reviews in eukaryotic gene expressionGonadal dysfunction and beyond: Clinical challenges in children, adolescents, and adults with 47,XXY Klinefelter syndrome.
American journal of medical genetics. Part C, Seminars in medical geneticsPituitary hyperplasia with Sertoli cell-only and 47,XYY syndromes: an uncommon triad.
BMJ case reportsClinical experience regarding the accuracy of NIPT in the detection of sex chromosome abnormality.
The journal of gene medicineNeurodevelopmental outcome of prenatally diagnosed boys with 47,XXY (Klinefelter syndrome) and the potential influence of early hormonal therapy.
American journal of medical genetics. Part AA patient with 46,XY/47,XYY karyotype and female phenotype: a case report.
BMC endocrine disordersMicrospherophakia in a 47, XYY Syndrome Patient: A Case Report.
Case reports in ophthalmologyImpact of early diagnosis and noninvasive prenatal testing (NIPT): Knowledge, attitudes, and experiences of parents of children with sex chromosome aneuploidies (SCAs).
Prenatal diagnosisDonor-derived XYY After BMT From a Healthy Volunteer.
TransplantationHigh false-positive non-invasive prenatal screening results for sex chromosome abnormalities: Are maternal factors the culprit?
Prenatal diagnosisAbnormal Auditory Mismatch Fields in Children and Adolescents with 47,XYY Syndrome.
Developmental neuroscienceCognitive Profile, Emotional-Behavioral Features, and Parental Stress in Boys With 47,XYY Syndrome.
Cognitive and behavioral neurology : official journal of the Society for Behavioral and Cognitive NeurologyAuditory evoked response delays in children with 47,XYY syndrome.
NeuroreportSensory Features as a Marker of Autism Spectrum Disorders.
Journal of autism and developmental disordersChanges in the cohort composition of turner syndrome and severe non-diagnosis of Klinefelter, 47,XXX and 47,XYY syndrome: a nationwide cohort study.
Orphanet journal of rare diseasesSelective advantage of euploid spermatocytes I in an azoospermic 47,XYY man with gonadal mosaicism.
Human reproduction (Oxford, England)Characterization of autism spectrum disorder and neurodevelopmental profiles in youth with XYY syndrome.
Journal of neurodevelopmental disordersSex differences in psychiatric disorders: what we can learn from sex chromosome aneuploidies.
Neuropsychopharmacology : official publication of the American College of NeuropsychopharmacologyOutcomes of Preimplantation Genetic Diagnosis Cycles by Fluorescent In situ Hybridization of Infertile Males with Nonmosaic 47,XYY Syndrome.
Chinese medical journalSurvival in double aneuploidy involving trisomy 18 and sex chromosome trisomy: A case report of a 27-month-old child and a review of the literature.
Congenital anomaliesBenefit of routine testicular examination: hypogonadism in a person with 47XYY.
Internal medicine journalCataract in a patient with 47,XYY sex chromosome aneuploidy.
Archivos de la Sociedad Espanola de OftalmologiaFertile offspring from sterile sex chromosome trisomic mice.
Science (New York, N.Y.)Sex chromosome aneuploidies and copy-number variants: a further explanation for neurodevelopmental prognosis variability?
European journal of human genetics : EJHGNoninvasive prenatal screening for fetal common sex chromosome aneuploidies from maternal blood.
The Journal of international medical researchDosage of Sex Chromosomal Genes in Blood Deposited on Filter Paper for Neonatal Screening of Sex Chromosome Aneuploidy.
Genetic testing and molecular biomarkersA boy with conduct disorder (CD), attention deficit hyperactivity disorder (ADHD), borderline intellectual disability, and 47,XXY syndrome in combination with a 7q11.23 duplication, 11p15.5 deletion, and 20q13.33 deletion.
Child and adolescent psychiatry and mental health[Genetic analysis of a child with XYY syndrome mainly featuring mental retardation].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsKlinefelter syndrome and 47,XYY syndrome in children with B cell acute lymphoblastic leukaemia.
British journal of haematologyPregnancy outcomes in prenatally diagnosed 47, XXX and 47, XYY syndromes: a 30-year French, retrospective, multicentre study.
Prenatal diagnosisXYY syndrome: a 13-year-old boy with tall stature.
Annals of pediatric endocrinology & metabolismRare genomic rearrangement in a boy with Williams-Beuren syndrome associated to XYY syndrome and intriguing behavior.
American journal of medical genetics. Part AOral, physical, and behavioral aspects of patient with chromosome 47, XYY syndrome.
Journal of the Indian Society of Pedodontics and Preventive DentistryOnly a minority of sex chromosome abnormalities are detected by a national prenatal screening program for Down syndrome.
Human reproduction (Oxford, England)Immature teratoma and subsequent acute promyelocytic leukemia in a pediatric patient with XYY syndrome.
Annals of laboratory medicine[Morphology and pathogenesis of 47, XYY/47, XY, +mar identified in patients with super male syndrome].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsPositive effects of early androgen therapy on the behavioral phenotype of boys with 47,XXY.
American journal of medical genetics. Part C, Seminars in medical geneticsA case of 45,X/47,XYY mosaicism in a male fetus with a hypoplastic nasal bone.
Journal of ultrasound in medicine : official journal of the American Institute of Ultrasound in MedicineIs microcephaly a so-far unrecognized feature of XYY syndrome?
Meta geneBehavioral phenotypes in males with XYY and possible role of increased NLGN4Y expression in autism features.
Genes, brain, and behaviorAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome 47,XYY.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome 47,XYY
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Neuropsychiatric Phenotype and Treatment Challenges in 47,XYY Syndrome: A Narrative Review with a Case Series of Adolescents.
- Neurodevelopmental and Mental Health Outcomes in a National Clinical Sample of Youth With Sex Chromosome Trisomies Compared With Matched Controls.
- Can Individuals with 47,XYY Karyotypes Exist without Male Phenotype? A Narrative Literature Review and Case Report.
- [Clinical characteristics and genetic analysis of a case with 47,XYY Disorder of sex development due to variant of NR5A1 gene].Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics· 2025· PMID 41070646mais citado
- An iPSC-based model of 47,XYY Jacobs syndrome reveals a DNA methylation-independent transcriptional dysregulation shared with male X aneuploid cells.
- Preimplantation genetic testing might not be the necessity for male patients with 47,XYY syndrome: A pilot study.
- Cytogenetic analysis of 3488 patients with recurrent pregnancy loss: An experience of two decades from a tertiary care center in South India.
- Gonadal function in patients with 47,XYY syndrome: a systematic review and meta-analysis.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:8(Orphanet)
- MONDO:0019339(MONDO)
- GARD:5674(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q267602(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
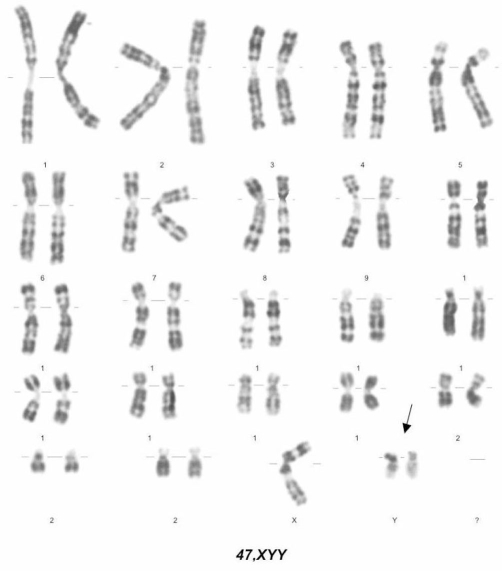
Síndrome 47,XYY
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov