Uma síndrome genética que causa múltiplas alterações congênitas (presentes desde o nascimento) e deficiência intelectual. Ela é provocada pela falta de um pedaço (deleção parcial) do braço longo do cromossomo 11.
Introdução
O que você precisa saber de cara
Uma síndrome genética que causa múltiplas alterações congênitas (presentes desde o nascimento) e deficiência intelectual. Ela é provocada pela falta de um pedaço (deleção parcial) do braço longo do cromossomo 11.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 30 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 105 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Not applicable, Unknown.
Sequence-specific transcriptional activator (PubMed:24100448, PubMed:26316623, PubMed:28255014). Recognizes the DNA sequence 5'-C[CA]GGAAGT-3'
Nucleus
Ewing sarcoma
A highly malignant, metastatic, primitive small round cell tumor of bone and soft tissue that affects children and adolescents. It belongs to the Ewing sarcoma family of tumors, a group of morphologically heterogeneous neoplasms that share the same cytogenetic features. They are considered neural tumors derived from cells of the neural crest. Ewing sarcoma represents the less differentiated form of the tumors.
Variantes genéticas (ClinVar)
108 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Jacobsen
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
De novo mutation in the ARHGAP32 gene endorses the implication of GTPase-activating proteins (RhoGAP family) in idiopathic autism spectrum disorder.
ARHGAP32 gene (Rho GTPase Activating Protein 32) encodes a Rho GTPase activating protein, which is vital for the regulation of synaptic plasticity and cytoskeletal dynamics. ARHGAP32 (11q24.3) has been implicated as a candidate gene for Autism Spectrum Disorder (ASD) in Jacobsen syndrome, where a 243-kb terminal deletion encompasses its locus. A unique patient with de novo (DN) likely gene-disruptive mutation of ARHGAP32 has been reported so far in the medical literature. The present study was undertaken to understand clinical, molecular, and neurobehavioral characteristics of ASD associated with a novel DN nonsense mutation in ARHGAP32. Clinical characterization included basal and follow-up assessment with standardized measures and comorbidities diagnosis. Trio exome sequencing analyses (WES) and variants annotation were performed. WES analyses of a 6-year-old female patient with idiopathic ASD revealed DN heterozygous nonsense variant in ARHGAP32 (NM_001378024.1: c.610C>T; NP_001364953.1: p.(Arg204Ter). The variant is predicted to introduce a premature stop codon, resulting in either a truncated protein or activation of nonsense-mediated mRNA decay, ultimately leading to loss of function. The patient presented with normative growth parameters and cranial measurements, with no congenital morphological anomalies. A diagnosis of idiopathic ASD was made at age 2. Developmental delays were observed, notably language regression beginning at 18 months, mild intellectual disability, and restricted interests accompanied by repetitive motor and verbal behaviors. Significant hyperactivity and attentional difficulties were observed. Over time, she exhibited borderline non-verbal cognitive functioning, persistent speech impairment, and was subsequently diagnosed with comorbid Attention Deficit Hyperactivity Disorder. This study identifies shared neurobehavioral features of idiopathic Autism Spectrum Disorder (ASD) associated with de novo LoF mutations in ARHGAP32 and reinforces the involvement of RhoGAP family proteins in neurodevelopmental disorders. Taken together with previous evidence, our data support the role of ARHGAP32 as a candidate gene for ASD, expanding the genetic spectrum.
A case with a de novo chromosome 8.9 Mb 11pter duplication and 6.4 Mb 11qter deletion derived from a father with a normal karyotype.
To determine the cause of marked hypotonia and neonatal encephalopathy, mild anemia and thrombocytopenia, and nonspecific facial dysmorphism in a neonate after extensive neurological and biochemical evaluations, and karyotyping failed to establish the etiology. Whole exome sequencing (WES), single-nucleotide polymorphisms chromosomal microarray (SNPs CMA), and fluorescence in-situ hybridization (FISH) analysis were performed in the patient and the parents. WES did not show a candidate gene/diagnosis. Trio-CMA and FISH confirmed a de novo 8.9 Mb duplication of 11p15.4p15.5 and a 6.4 Mb deletion of 11q24.3q25 in the child, and that the duplicated 11p was of paternal origin, based on the SNPs analysis. On long-term follow-up, encephalopathy improved gradually, thrombocytopenia resolved, facial puffiness persisted, and developmental delay remained. The present patient represents the first case of de novo 11p duplication with 11q deletion and severe neonatal encephalopathy. The de novo chromosomal rearrangement could still have resulted from nonallelic homologous recombination, triggered by low-copy repeats (LCRs) at distal 11p and 11q during paternal meiosis, despite the lower LCR density. Alternatively, it may represent a random event, as it does not involve the recurrent deletion/duplication typically observed in regions with dense LCRs.
Ets1-regulated endothelial-secreted factors promote compact myocardial growth and contribute to the pathogenesis of ventricular non-compaction.
Thinning of the compact myocardium is a major contributor to adverse outcomes in ventricular non-compaction, the third most common form of cardiomyopathy. Endothelial-specific deletion of Ets1, a gene associated with Jacobsen syndrome, causes ventricular non-compaction with reduced compact myocardium. However, the mechanisms by which pathological cardiac endothelium impairs compact myocardium growth remain poorly understood. To uncover the mechanisms underlying compact myocardium thinning and identify therapeutic endothelial-secreted factors, we performed single-cell RNA sequencing. Aberrant cardiomyocyte and endothelial cell states were observed in non-compacted ventricles. Conditional deletion of Ets1 in either the endocardium or coronary endothelium impaired compact myocardial growth. In endocardium, Ets1 deficiency suppressed Notch1 signaling by upregulating Dlk1 and downregulating Dll4, both direct Ets1 targets. In coronary endothelium, Ets1 deficiency reduced the expression of its direct targets Hmcn1, Slit2, and Col18a1, three extracellular matrix (ECM) components that promote compact myocardial proliferation. Notably, treatment with these ECM proteins or the Notch1 effector Nrg1 restored the impaired compact myocardial proliferation. These findings highlight Ets1-regulated endothelial-secreted factors as essential for compact myocardium development and suggest novel therapeutic targets for ventricular non-compaction.
Evaluation of the synapse adhesion molecule Kirrel3 in neurological disease.
The synaptic adhesion molecule KIRREL3 regulates synapse development in mice and is implicated in human neurological disorders, including autism spectrum disorder, intellectual disability, and Jacobsen syndrome (chromosome 11q deletion syndrome). However, its status as a definitive human disease gene remains unresolved, likely due to the rarity of KIRREL3-related disorders and significant gaps in understanding its molecular mechanisms. Current knowledge is further fragmented across disparate clinical and basic research reports, often buried in supplemental data. This review synthesizes existing evidence to enable clinicians and scientists to better evaluate KIRREL3 variants as potentially disease causing. We review its conserved role in mediating neuron-to-neuron interactions during axon targeting and synapse formation in mice and how disruptions to these interactions could contribute to neurological pathology in humans. We also discuss how disease-associated variants alter KIRREL3 function. Our analysis underscores the need for integrated studies spanning basic and clinical investigation to validate KIRREL3's disease association and advance future interventions for KIRREL3-related disorders.
Clinical and neuropsychological characterization of Jacobsen syndrome (del11q).
Jacobsen Syndrome (JS), also known as 11q Deletion Syndrome (del11q), is a rare genetic disorder affecting approximately 1 in 100,000 births that presents with varied clinical manifestations and severities including intellectual disability, psychomotor delays, and distinctive physical traits. This study offers a detailed analysis of the clinical and cognitive profiles of individuals with JS and examines how these characteristics are related to each other and to genetic variables. Twenty-nine participants with JS (20 female, mean age 12.48 years, SD = 9.13) underwent standardized assessments assessing cognitive functioning, adaptive behavior, autistic traits, and general psychopathology. A CGH array was used to assess genetic deletions. We employed descriptive and inferential statistical analyses to explore the association between clinical and cognitive characteristics and deletion size. Sixty percent of participants had verbal language. Mean intelligence quotient was 50.18, the range of adaptive functioning was very broad, and 43% showed behaviors exceeding the ADOS-2 cutoff for autism spectrum classification. A higher cognitive performance was associated with better adaptive skills, including more advanced language skills and with more depressive symptoms or a diagnosis of depression. Larger deletions were associated with more delays in developmental milestones and poorer cognitive functioning. No significant association was found between haploinsufficiency of the KIRREL3 and ARHGAP32 genes and cognitive functioning or autistic characteristics. Our findings provide deeper insights into the complex relationship between genetic factors and clinical attributes in individuals with JS, revealing notable clinical variability within the JS population. This information may help predict developmental difficulties as genetic findings emerge.
Publicações recentes
De novo mutation in the ARHGAP32 gene endorses the implication of GTPase-activating proteins (RhoGAP family) in idiopathic autism spectrum disorder.
🥈 Ensaio clínicoA case with a de novo chromosome 8.9 Mb 11pter duplication and 6.4 Mb 11qter deletion derived from a father with a normal karyotype.
Clinical and neuropsychological characterization of Jacobsen syndrome (del11q).
Ets1-regulated endothelial-secreted factors promote compact myocardial growth and contribute to the pathogenesis of ventricular non-compaction.
Jacobsen Syndrome With White Matter Abnormalities: A Case Report and MRI Follow-Up.
📚 EuropePMC123 artigos no totalmostrando 91
De novo mutation in the ARHGAP32 gene endorses the implication of GTPase-activating proteins (RhoGAP family) in idiopathic autism spectrum disorder.
Frontiers in psychiatryA case with a de novo chromosome 8.9 Mb 11pter duplication and 6.4 Mb 11qter deletion derived from a father with a normal karyotype.
Clinical dysmorphologyClinical and neuropsychological characterization of Jacobsen syndrome (del11q).
Journal of neurodevelopmental disordersEts1-regulated endothelial-secreted factors promote compact myocardial growth and contribute to the pathogenesis of ventricular non-compaction.
Cardiovascular researchJacobsen Syndrome With White Matter Abnormalities: A Case Report and MRI Follow-Up.
CureusJacobsen Syndrome: A Case Report With Olfactory Bulb Agenesis, Severe Endocrinopathy, and Neurodevelopmental Delay.
Clinical case reportsEvaluation of the synapse adhesion molecule Kirrel3 in neurological disease.
Frontiers in neurologyKaposi-Juliusberg syndrome in a young man with Jacobsen syndrome.
International journal of infectious diseases : IJID : official publication of the International Society for Infectious DiseasesJacobsen Syndrome, Paris-Trousseau Syndrome, and Dental Extractions: Case Report, Medical, Dental, and Social Considerations.
Special care in dentistry : official publication of the American Association of Hospital Dentists, the Academy of Dentistry for the Handicapped, and the American Society for Geriatric DentistryPrenatal Diagnosis of Unbalanced Translocation t(5;11)(q21;q22) With a Jacobsen Syndrome Clinical Phenotype.
Clinical case reportsFamilial Exudative Vitreoretinopathy in a Patient with Jacobsen Syndrome: A Case Report.
Korean journal of ophthalmology : KJOInterstitial 11q Deletions and Terminal 11q Duplications Cause a Bleeding Tendency due to Platelet Dysfunction That Is Similar to 11q Deletions Causing Jacobsen Syndrome.
European journal of haematologyJacobsen syndrome associated with Shone's complex: a case report.
Revista paulista de pediatria : orgao oficial da Sociedade de Pediatria de Sao PauloPrenatal diagnosis of Jacobsen syndrome associated with a distal 11q deletion and a distal 8q duplication by chromosome microarray analysis in a fetus with a de novo unbalanced translocation of 46,XX,der(11)t(8;11)(q24.13;q23.3) and multiple congenital anomalies on fetal ultrasound.
Taiwanese journal of obstetrics & gynecologyHuman Genetics of Tetralogy of Fallot and Double-Outlet Right Ventricle.
Advances in experimental medicine and biologyHuman Genetics of Ventricular Septal Defect.
Advances in experimental medicine and biologyA bird's eye view on the use of whole exome sequencing in rare congenital ophthalmic diseases.
Journal of human geneticsFEZ1 participates in human embryonic brain development by modulating neuronal progenitor subpopulation specification and migrations.
iScienceRecurrent pneumonia in a child with Jacobsen syndrome and common variable immune deficiency.
Clinical case reportsHematological abnormalities in Jacobsen syndrome: cytopenia of varying severities and morphological abnormalities in peripheral blood and bone marrow.
HaematologicaJacobsen Syndrome in a Patient with Combined Immunodeficiency, Thrombocytopenia, and Lymphoma.
Journal of investigational allergology & clinical immunologyJacobsen syndrome: Chromosome deletion at llq23.
Journal of osteopathic medicineJacobsen Syndrome with Hypoplastic Left Heart Syndrome: Outcome after Cardiac Transplantation.
Journal of cardiovascular development and diseasePrenatal Identification and Confirmation of Jacobsen Syndrome: A Series of Four Cases.
Journal of the College of Physicians and Surgeons--Pakistan : JCPSPCase report: ETS1 gene deletion associated with a low number of recent thymic emigrants in three patients with Jacobsen syndrome.
Frontiers in immunologyDetailed analysis of mortality rates in the female progeny of 1,001 Holstein bulls allows the discovery of new dominant genetic defects.
Journal of dairy scienceDeletion of 11q24.2-qter in a male child with cleft lip and palate: an atypical feature of Jacobsen syndrome.
Journal of geneticsUtility of thromboelastogram in cardiac surgery in Jacobsen syndrome associated with platelet dysfunction: a case report.
JA clinical reportsETS1 loss in mice impairs cardiac outflow tract septation via a cell migration defect autonomous to the neural crest.
Human molecular geneticsEndothelial Loss of ETS1 Impairs Coronary Vascular Development and Leads to Ventricular Non-Compaction.
Circulation researchETS1 and HLHS: Implications for the Role of the Endocardium.
Journal of cardiovascular development and diseasePatients with Chromosome 11q Deletions Are Characterized by Inborn Errors of Immunity Involving both B and T Lymphocytes.
Journal of clinical immunologyFirst Report of Jacobsen Syndrome with Dextrocardia Diagnosed with del(11)(q24q25).
Molecular syndromologyNeonatal interstitial lung disease in a girl with Jacobsen syndrome: a case report.
Journal of medical case reportsSpectrum of Genetic T-Cell Disorders from 22q11.2DS to CHARGE.
Clinical reviews in allergy & immunologyImmunodeficiency and Lymphoma in Jacobsen Syndrome.
Journal of investigational allergology & clinical immunologyJacobsen syndrome with bilateral periventricular white matter lesions.
World journal of pediatrics : WJPImmunological Evaluation of Patients Affected with Jacobsen Syndrome Reveals Profound Not Age-Related Lymphocyte Alterations.
Journal of clinical immunologyRecurrent Spontaneous Intracranial Hemorrhage in a Patient With Jacobsen Syndrome.
Neurology. Clinical practiceImmune Deficiency in Jacobsen Syndrome: Molecular and Phenotypic Characterization.
GenesJacobsen syndrome and neonatal bleeding: report on two unrelated patients.
Italian journal of pediatricsAlteration of the Arcuate Fasciculus in Jacobsen Syndrome Shown by Diffusion Tensor Imaging.
Pediatric neurologyLoss of FEZ1, a gene deleted in Jacobsen syndrome, causes locomotion defects and early mortality by impairing motor neuron development.
Human molecular geneticsTen-year use of recombinant parathyroid hormone for the treatment of hypoparathyroidism in a boy with partial Jacobsen syndrome.
Pediatric endocrinology, diabetes, and metabolismTopical cidofovir for the treatment of recalcitrant viral warts and molluscum contagiosum in Jacobsen syndrome.
Pediatric dermatologyChromoanasynthesis as a cause of Jacobsen syndrome.
American journal of medical genetics. Part AMultiple cerebral cysts are another possible feature of Jacobsen syndrome.
Brain & developmentWhite matter abnormality in Jacobsen syndrome assessed by serial MRI.
Brain & developmentReader response: Teaching NeuroImages: A rare case of Jacobsen syndrome with global diffuse hypomyelination of brain.
NeurologyUltrasonographic findings and prenatal diagnosis of Jacobsen syndrome: A case report and review of the literature.
MedicineScurvy Findings in a Child with Jacobsen Syndrome: A Case Report.
JBJS case connector[Prenatal diagnosis of Jacobsen syndrome in a fetus carried by a pregnant woman with intellectual disability].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsPartial Jacobsen syndrome phenotype in a patient with a de novo frameshift mutation in the ETS1 transcription factor.
Cold Spring Harbor molecular case studiesObstructive Sleep Apnea in Jacobsen Syndrome.
Sleep and vigilanceSuccessful Management of a Patient With Jacobsen Syndrome and Hypoplastic Left Heart Syndrome.
World journal for pediatric & congenital heart surgeryTeaching NeuroImages: A rare case of Jacobsen syndrome with global diffuse hypomyelination of brain.
Neurology11q24.2q24.3 microdeletion in two families presenting features of Jacobsen syndrome, without intellectual disability: Role of FLI1, ETS1, and SENCR long noncoding RNA.
American journal of medical genetics. Part AHypoplastic Left Heart Syndrome: A New Paradigm for an Old Disease?
Journal of cardiovascular development and diseaseGene-targeted deletion in mice of the Ets-1 transcription factor, a candidate gene in the Jacobsen syndrome kidney "critical region," causes abnormal kidney development.
American journal of medical genetics. Part ANeph2/Kirrel3 regulates sensory input, motor coordination, and home-cage activity in rodents.
Genes, brain, and behaviorTwo siblings with 11qter deletion syndrome that had been rescued in their mother by uniparental disomy.
European journal of medical geneticsVentriculomegaly and cerebellar hypoplasia in a neonate with interstitial 11q 24 deletion in Jacobsen syndrome region.
Clinical case reportsCraniosynostosis as a clinical and diagnostic problem: molecular pathology and genetic counseling.
Journal of applied genetics11q23 deletion syndrome (Jacobsen syndrome) with severe bleeding: a case report.
Journal of medical case reportsFLI1 level during megakaryopoiesis affects thrombopoiesis and platelet biology.
BloodIncreased Excitatory Synaptic Transmission of Dentate Granule Neurons in Mice Lacking PSD-95-Interacting Adhesion Molecule Neph2/Kirrel3 during the Early Postnatal Period.
Frontiers in molecular neuroscienceMolecular cytogenetic characterization of Jacobsen syndrome (11q23.3-q25 deletion) in a fetus associated with double outlet right ventricle, hypoplastic left heart syndrome and ductus venosus agenesis on prenatal ultrasound.
Taiwanese journal of obstetrics & gynecologyComplex Mosaic Ring Chromosome 11 Associated with Hemizygous Loss of 8.6 Mb of 11q24.2qter in Atypical Jacobsen Syndrome.
Molecular syndromologyBrain hemorrhages in Jacobsen syndrome: A retrospective review of six cases and clinical recommendations.
American journal of medical genetics. Part AMorphological and genetic abnormalities in a Jacobsen syndrome.
Romanian journal of morphology and embryology = Revue roumaine de morphologie et embryologie11q terminal deletion and combined immunodeficiency (Jacobsen syndrome): Case report and literature review on immunodeficiency in Jacobsen syndrome.
American journal of medical genetics. Part AJacobsen syndrome, Braddock-Carey syndrome, and Beyond: Reflections on intellectual disability accompanied with thrombocytopenia.
American journal of medical genetics. Part AA case of Jacobsen syndrome with multifocal white matter lesions.
Clinical imagingde novo interstitial deletions at the 11q23.3-q24.2 region.
Molecular cytogeneticsJacobsen Syndrome: Surgical Complications due to Unsuspected Diagnosis, the Importance of Molecular Studies in Patients with Craniosynostosis.
Molecular syndromologyPX-RICS-deficient mice mimic autism spectrum disorder in Jacobsen syndrome through impaired GABAA receptor trafficking.
Nature communicationsEndocrine abnormalities in ring chromosome 11: a case report and review of the literature.
Endocrinology, diabetes & metabolism case reportsThe 11q Terminal Deletion Disorder Jacobsen Syndrome is a Syndromic Primary Immunodeficiency.
Journal of clinical immunologyMacrothrombocytopenia and developmental delay with a de novo CDC42 mutation: Yet another locus for thrombocytopenia and developmental delay.
American journal of medical genetics. Part A11q24.2-25 micro-rearrangements in autism spectrum disorders: Relation to brain structures.
American journal of medical genetics. Part AParis-Trousseau thrombocytopenia is phenocopied by the autosomal recessive inheritance of a DNA-binding domain mutation in FLI1.
BloodJacobsen syndrome: Advances in our knowledge of phenotype and genotype.
American journal of medical genetics. Part C, Seminars in medical geneticsMice lacking the synaptic adhesion molecule Neph2/Kirrel3 display moderate hyperactivity and defective novel object preference.
Frontiers in cellular neuroscience[Prenatal diagnosis of a case with combined Wolf-Hirschhorn syndrome and Jacobsen syndrome].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsLeukoencephalopathy associated with 11q24 deletion involving the gene encoding hepatic and glial cell adhesion molecule in two patients.
European journal of medical geneticsMice Haploinsufficient for Ets1 and Fli1 Display Middle Ear Abnormalities and Model Aspects of Jacobsen Syndrome.
The American journal of pathologyA Case Report: Jacobsen Syndrome Complicated by Paris-Trousseau Syndrome and Shone's Complex.
Journal of pediatric hematology/oncologyAutism and Intellectual Disability-Associated KIRREL3 Interacts with Neuronal Proteins MAP1B and MYO16 with Potential Roles in Neurodevelopment.
PloS onePartial monosomy of 11q22.2q22.3 including the SDHD gene in individuals with developmental delay.
American journal of medical genetics. Part AClinical and molecular evaluations of siblings with "pure" 11q23.3-qter trisomy or reciprocal monosomy due to a familial translocation t (10;11) (q26;q23.3).
Molecular cytogeneticsJacobsen syndrome detected by noninvasive prenatal testing.
Obstetrics and gynecologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Jacobsen.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Jacobsen
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- De novo mutation in the ARHGAP32 gene endorses the implication of GTPase-activating proteins (RhoGAP family) in idiopathic autism spectrum disorder.
- A case with a de novo chromosome 8.9 Mb 11pter duplication and 6.4 Mb 11qter deletion derived from a father with a normal karyotype.
- Ets1-regulated endothelial-secreted factors promote compact myocardial growth and contribute to the pathogenesis of ventricular non-compaction.
- Evaluation of the synapse adhesion molecule Kirrel3 in neurological disease.
- Clinical and neuropsychological characterization of Jacobsen syndrome (del11q).
- Jacobsen Syndrome With White Matter Abnormalities: A Case Report and MRI Follow-Up.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2308(Orphanet)
- OMIM OMIM:147791(OMIM)
- MONDO:0007838(MONDO)
- GARD:307(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q1677755(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
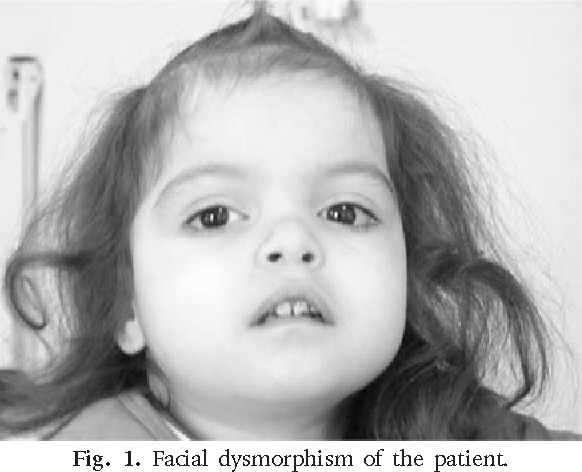
Síndrome Jacobsen
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov