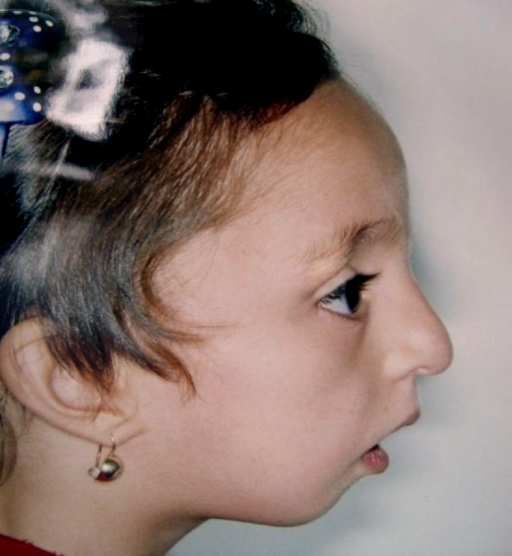
A Síndrome de Nager, também conhecida como disostose acrofacial de Nager (NAFD), é uma condição de má-formação congênita (presente desde o nascimento). Ela é caracterizada por alterações no desenvolvimento do rosto e da mandíbula, como maçãs do rosto subdesenvolvidas (hipoplasia malar), queixo pequeno (micrognatia) e má-formações nas orelhas externas. Também apresenta, em graus variados, defeitos nos membros (braços e pernas), especialmente na parte que corresponde ao polegar ou dedão do pé.
Introdução
O que você precisa saber de cara
A Síndrome de Nager, também conhecida como disostose acrofacial de Nager (NAFD), é uma condição de má-formação congênita (presente desde o nascimento). Ela é caracterizada por alterações no desenvolvimento do rosto e da mandíbula, como maçãs do rosto subdesenvolvidas (hipoplasia malar), queixo pequeno (micrognatia) e má-formações nas orelhas externas. Também apresenta, em graus variados, defeitos nos membros (braços e pernas), especialmente na parte que corresponde ao polegar ou dedão do pé.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 31 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 98 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive, Not applicable.
Curadoria gene-doença
fontes oficiaisComponent of the 17S U2 SnRNP complex of the spliceosome, a large ribonucleoprotein complex that removes introns from transcribed pre-mRNAs (PubMed:10882114, PubMed:12234937, PubMed:27720643, PubMed:32494006). The 17S U2 SnRNP complex (1) directly participates in early spliceosome assembly and (2) mediates recognition of the intron branch site during pre-mRNA splicing by promoting the selection of the pre-mRNA branch-site adenosine, the nucleophile for the first step of splicing (PubMed:12234937
Nucleus
Acrofacial dysostosis 1, Nager type
A form of acrofacial dysostosis, a group of disorders which are characterized by malformation of the craniofacial skeleton and the limbs. The major facial features of AFD1 include downslanted palpebral fissures, midface retrusion, and micrognathia, the latter of which often requires the placement of a tracheostomy in early childhood. Limb defects typically involve the anterior (radial) elements of the upper limbs and manifest as small or absent thumbs, triphalangeal thumbs, radial hyoplasia or aplasia, and radioulnar synostosis. Phocomelia of the upper limbs and, occasionally, lower-limb defects have also been reported.
Variantes genéticas (ClinVar)
69 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 39 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
4 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Nager
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
Pesquisa e ensaios clínicos
1 ensaios clínicos encontrados.
Publicações mais relevantes
Nager Syndrome Revisited: Integrating In Vivo and In Vitro Models to Decipher SF3B4-Dependent Tissue Coordination.
Nager syndrome (NS) is a rare congenital disorder primarily characterized by mandibulofacial dysostosis and upper limb anomalies. Pathogenic variants in SF3B4, which encodes a core spliceosomal component, represent the primary known genetic cause of NS. This review synthesizes recent findings from cellular, zebrafish, Xenopus, and mouse models to elucidate how SF3B4 deficiency perturbs neural crest cell (NCC) biology and multi-tissue development. Loss of SF3B4 induces widespread splicing abnormalities, with preferential exon skipping affecting AT-rich and GC-poor exons, thereby altering the expression of genes critical for NCC survival, proliferation, migration, and lineage specification. These cellular defects are further exacerbated by oxidative stress and activation of the p53 pathway, resulting in a broad spectrum of developmental abnormalities involving craniofacial, cardiac, skeletal, and sensory (auditory and ocular) systems. Together, these findings highlight the essential role of SF3B4 in coordinating early morphogenesis. Cross-species comparisons reveal conserved NCC vulnerabilities alongside model-specific phenotypes, highlighting the challenge of linking individual splicing alterations to distinct structural outcomes in NS. Future research directions include defining tissue-specific SF3B4-dependent splicing targets, developing human induced pluripotent stem cell-derived models, and exploring therapeutic strategies aimed at restoring splicing homeostasis or compensating for disrupted developmental signaling pathways. This article is categorized under: Congenital Diseases > Molecular and Cellular Physiology Congenital Diseases > Genetics/Genomics/Epigenetics Congenital Diseases > Stem Cells and Development.
Management and Outcomes of Neonates with Treacher Collins and Nager Syndromes.
To compare management and outcomes of infants with mandibulofacial dysostosis syndromes (Treacher Collins and Nager syndromes) admitted to neonatal intensive care units (NICUs) to infants with other causes of micrognathia. The Children's Hospitals Neonatal Database from 2010 to 2023 was queried for infants with diagnoses of Treacher Collins syndrome (TCS), Nager syndrome (NS), and other infants in NICUs with micrognathia (n = 4210). We identified 103 infants with TCS and 11 with NS to compare with the micrognathia cohort (n = 4210). Compared with infants with micrognathia, those with TCS were more likely to undergo tracheostomy (54% vs 11%) and gastrostomy tube placement (67% vs 35%) and were less likely to undergo mandibular distraction (9.7% vs 28.2%). The hospital mortality rate in TCS was lower than micrognathia cohort (1.9% vs 7.2%). Apgar scores were similar for TCS and micrognathia cohorts (6 and 8 vs 7 and 8, at 1 and 5 minutes, respectively) but lower for NS (2 and 6). Infants with NS had the highest rate of intubation at birth (91%) and tracheostomy placement (72.7%), and a higher mortality rate than TCS (27.3% vs 1.9%). Hospital length of stay was longer in TCS (47.5 days) and NS (43 days) than the micrognathia cohort (37 days). Infants with mandibulofacial dysostosis (TCS and NS) were more likely to have a tracheostomy and gastrostomy tube, and less likely to undergo mandibular distraction than infants with micrognathia from other causes. NS was most severe with highest mortality rate and lowest Apgar scores. Despite a higher rate of tracheostomy and longer length of stay, the mortality rate for TCS remained low.
Transcriptome-wide profiling of alternative splicing regulators with CRISPore-seq.
Alternative splicing creates diverse RNA isoforms from individual genes, yet single-cell CRISPR screens are limited to gene-level quantification and cannot detect changes in alternative splicing and transcript isoforms. To overcome this limitation, we develop CRISPore-seq, which couples massively-parallel CRISPR perturbations with joint short- and long-read transcriptomics. CRISPore-seq simultaneously captures genetic perturbations and expression of genes, full-length transcripts and surface proteins in single cells. CRISPore-seq long reads identify 80% more transcript isoforms than short reads. Nearly all long reads map to unique transcript isoforms - in contrast to existing single-cell perturbation methods, which rarely distinguish specific isoforms. Using CRISPore-seq, we knock-down 15 different RNA-binding proteins (RBPs) and identify thousands of perturbation-driven alternative splicing events (ASEs). We find that exon skipping is the most common ASE observed and that skipped exons are enriched for binding sites of perturbed RBPs. Loss of the Nager syndrome-associated spliceosomal factor SF3B4 triggers skipping of exon 2 in the cell-cycle regulator CCND1 , preventing formation of a complex with CDK6 and blocking the G1-S transition. After rescue with a CCND1 isoform containing the skipped exon, both holoenzyme complex formation and cell proliferation are restored. By linking genes to transcriptional phenotypes with isoform-level resolution, CRISPore-seq is a highly scalable tool for understanding the impact of genetic perturbations on the human transcriptome.
A New Case of Nager Syndrome as a Rare Cause of Acrofacial Dysostosis.
Nager acrofacial dysostosis (#154400) is a rare and mostly sporadic malformation syndrome characterized by craniofacial and extremity findings. In the study, a new case diagnosed in the neonatal period will be presented. The neonatal intensive care unit consulted with our pediatrics genetic clinic for a 2-week-old male patient, who was being followed up in their unit, due to his dysmorphic findings and extremity defects. In the physical examination, downward palpebral fissures, zygomatic bone hypoplasia, mandibular hypoplasia, retromicrognathia, bilateral microtia, bilateral external auditory canal atresia, and bilateral thumb agenesis were observed. Direct radiographs showed left radius hypoplasia and bilateral thumb agenesis. Nager syndrome was considered in the presence of typical craniofacial findings, preaxial extremity deformities, and radiological findings. In the SF3B4 sequence analysis, c.737dupC p.(V247Sfs*239) heterozygous variant was detected. As a result of the segregation analysis, it was demonstrated that the variant was de novo. Nager acrofacial dysostosis should be considered in the patients with craniofacial malformations and radial ray findings.
PTBP3 Associated With 9q32 Locus Is a Candidate Gene for Nager Syndrome.
Mandibulofacial dysostosis (MFD) is a congenital disorder characterized by defects in facial bones of neural crest origin. Nager syndrome combines many features of MFD with limb defects. Mutations in SF3B4, a gene located on chromosome 1 that encodes a protein of the spliceosome, were identified as a cause for Nager syndrome in approximately 60% of patients. A region of chromosome 9 (9q32) that contains 35 genes has also been linked to Nager syndrome and may account for some affected individuals for which the causes of the disease have not been identified. Because Nager syndrome belongs to a rapidly growing list of craniofacial syndromes caused by pathogenic variants of splicing factors, we focused our attention on two genes in the 9q32 region that encode factors involved in pre-mRNA processing, PRPF4 and PTBP3, and analyzed their role in craniofacial development in Xenopus embryos. Loss-of-function experiments in Xenopus laevis embryos indicate that Ptbp3 is required while Prpf4 is dispensable for neural crest gene expression at the neurula stage, a phenotype that is partially rescued by expression of human PTBP3. At the tadpole stage, Ptbp3-depleted embryos have severely hypoplastic craniofacial cartilages, phenocopying the defects observed in Sf3b4 morphant tadpoles. Furthermore, ptbp3 expression is significantly increased in Xenopus tropicalis sf3b4 Null embryos as compared to wild-type siblings. We propose that dysregulation of PTBP3 expression may cause Nager syndrome in a subset of patients who do not have a mutation in SF3B4.
Publicações recentes
Nager Syndrome Revisited: Integrating In Vivo and In Vitro Models to Decipher SF3B4-Dependent Tissue Coordination.
Transcriptome-wide profiling of alternative splicing regulators with CRISPore-seq.
A New Case of Nager Syndrome as a Rare Cause of Acrofacial Dysostosis.
PTBP3 Associated With 9q32 Locus Is a Candidate Gene for Nager Syndrome.
The Development of a European Registry for Facial Dysostosis Syndromes: A Delphi-Guided Approach.
📚 EuropePMC68 artigos no totalmostrando 59
Nager Syndrome Revisited: Integrating In Vivo and In Vitro Models to Decipher SF3B4-Dependent Tissue Coordination.
WIREs mechanisms of diseaseTranscriptome-wide profiling of alternative splicing regulators with CRISPore-seq.
bioRxiv : the preprint server for biologyA New Case of Nager Syndrome as a Rare Cause of Acrofacial Dysostosis.
Molecular syndromologyPTBP3 Associated With 9q32 Locus Is a Candidate Gene for Nager Syndrome.
Birth defects researchThe Development of a European Registry for Facial Dysostosis Syndromes: A Delphi-Guided Approach.
The Journal of craniofacial surgeryIn silico analysis identified potential interaction between glutathione and spliceosome in Nager Syndrome.
Free radical biology & medicineUnveiling the Phenotypic Spectrum of Miller Syndrome: A Systematic Review.
The Journal of craniofacial surgeryManagement and Outcomes of Neonates with Treacher Collins and Nager Syndromes.
The Journal of pediatricsDeletion of sf3b4 causes splicing defects and gene dysregulation that disrupt craniofacial development and survival.
Disease models & mechanismsHuman stem cell model of neural crest cell differentiation reveals a requirement of SF3B4 in survival, maintenance, and differentiation.
Developmental dynamics : an official publication of the American Association of AnatomistsComplete Agenesis of Right Half of Soft Palate-A Case Report.
Indian journal of plastic surgery : official publication of the Association of Plastic Surgeons of IndiaFgf8 contributes to the pathogenesis of Nager syndrome.
International journal of biological macromoleculesTranscriptomic analysis reveals mitochondrial dysfunction in the pathogenesis of Nager syndrome in sf3b4-depleted zebrafish.
Biochimica et biophysica acta. Molecular basis of diseaseSf3b4 mutation in Xenopus tropicalis causes RNA splicing defects followed by massive gene dysregulation that disrupt cranial neural crest development.
bioRxiv : the preprint server for biologyChildren with Rare Nager Syndrome-Literature Review, Clinical and Physiotherapeutic Management.
GenesDe novo PHF5A variants are associated with craniofacial abnormalities, developmental delay, and hypospadias.
Genetics in medicine : official journal of the American College of Medical GeneticsDysregulation of Spliceosomes Complex Induces Retinitis Pigmentosa-Like Characteristics in sf3b4-Depleted Zebrafish.
The American journal of pathologySF3B4 promotes Twist1 expression and clear cell renal cell carcinoma progression by facilitating the export of KLF 16 mRNA from the nucleus to the cytoplasm.
Cell death & diseaseFirst case report of Nager syndrome patient from Georgia.
SAGE open medical case reportsSF3B4 Frameshift Variants Represented a More Severe Clinical Manifestation in Nager Syndrome.
The Cleft palate-craniofacial journal : official publication of the American Cleft Palate-Craniofacial AssociationSPINKs in Tumors: Potential Therapeutic Targets.
Frontiers in oncologyThe Role of Splicing Factor SF3B4 in Congenital Diseases and Tumors.
Discovery medicinePossible autosomal recessive inheritance in a neonate with Nager syndrome: Case report.
Annals of medicine and surgery (2012)Fetal Skeletal Dysplasias that Involve the Face: Binder Syndrome and Nager Syndrome.
MaedicaAlloplastic Temporomandibular Joint Reconstruction in Congenital Craniofacial Deformities.
The Journal of craniofacial surgeryMolecular mechanisms of hearing loss in Nager syndrome.
Developmental biology[Obstructive sleep apnea in microtia children with maxillofacial dysostosis].
Lin chuang er bi yan hou tou jing wai ke za zhi = Journal of clinical otorhinolaryngology head and neck surgeryThe role of double-step advancement genioplasty and bilateral coronoidectomy in Nager Syndrome: A case report.
Special care in dentistry : official publication of the American Association of Hospital Dentists, the Academy of Dentistry for the Handicapped, and the American Society for Geriatric DentistryNager syndrome in patient lacking acrofacial dysostosis: Expanding the phenotypic spectrum of SF3B4-related disease.
American journal of medical genetics. Part ATargeted Next-Generation Sequencing in the Diagnosis of Facial Dysostoses.
Frontiers in geneticsBroad-spectrum next-generation sequencing-based diagnosis of a case of Nager syndrome.
Journal of clinical laboratory analysisSecond-trimester prenatal diagnosis of Nager syndrome with a deletion including SF3B4 detected by chromosomal microarray.
Clinical case reportsSF3b4: A Versatile Player in Eukaryotic Cells.
Frontiers in cell and developmental biologyAssociated syndromes in patients with Pierre Robin Sequence.
International journal of pediatric otorhinolaryngologyHeterozygous mutation of the splicing factor Sf3b4 affects development of the axial skeleton and forebrain in mouse.
Developmental dynamics : an official publication of the American Association of AnatomistsA Case of Nager Syndrome Diagnosed Before Birth.
Acta medica OkayamaThe final demise of Rodriguez lethal acrofacial dysostosis: A case report and review of the literature.
American journal of medical genetics. Part AMicrotia and Related Facial Anomalies.
Clinics in perinatologyModified Lefort Distraction Osteogenesis for the Treatment of Nager Syndrome-Associated Midface Hypoplasia: Technique and Review.
The Journal of craniofacial surgeryDecannulation and Airway Outcomes With Maxillomandibular Distraction in Treacher Collins and Nager Syndrome.
The Journal of craniofacial surgeryCongenital Abnormalities of the Temporomandibular Joint.
Oral and maxillofacial surgery clinics of North AmericaAnkylosis of temporomandibular joints after mandibular distraction osteogenesis in patients with Nager syndrome: Report of two cases and literature review.
Journal of plastic, reconstructive & aesthetic surgery : JPRASSynchronous Bilateral Breast Cancer in a Patient With Nager Syndrome.
Clinical breast cancerA synonymous splicing mutation in the SF3B4 gene segregates in a family with highly variable Nager syndrome.
European journal of human genetics : EJHGDoes canal wall down mastoidectomy benefit syndromic children with congenital aural stenosis?
International journal of pediatric otorhinolaryngologyA Case Report of Absent Epiglottis in Children With Nager Syndrome: Its Impact on Swallowing.
The Cleft palate-craniofacial journal : official publication of the American Cleft Palate-Craniofacial AssociationRodriguez acrofacial dysostosis is caused by apparently de novo heterozygous mutations in the SF3B4 gene.
American journal of medical genetics. Part AAltered mRNA Splicing, Chondrocyte Gene Expression and Abnormal Skeletal Development due to SF3B4 Mutations in Rodriguez Acrofacial Dysostosis.
PLoS geneticsUse of the C-MAC® adult D blade in paediatric patients with Nager syndrome.
Anaesthesia and intensive careThe Craniofacial and Upper Limb Management of Nager Syndrome.
The Journal of craniofacial surgeryPropranolol-induced gingival hyperplasia with Nager syndrome: A rare adverse drug reaction.
Journal of advanced pharmaceutical technology & researchNager Syndrome with Eventration of Diaphragm: A Rare Presentation.
Indian journal of pediatricsSf3b4-depleted Xenopus embryos: A model to study the pathogenesis of craniofacial defects in Nager syndrome.
Developmental biologyPrenatal diagnosis of Nager syndrome in a 12-week-old fetus with a whole gene deletion of SF3B4 by chromosomal microarray.
European journal of medical geneticsDental Management of a Patient with Nager Acrofacial Dysostosis.
Case reports in dentistry[Nager syndrome associated with tetralogy of Fallot: A frequent association?].
Archives de pediatrie : organe officiel de la Societe francaise de pediatrieA review of craniofacial disorders caused by spliceosomal defects.
Clinical geneticsNager syndrome and Pierre Robin sequence.
Pediatrics international : official journal of the Japan Pediatric SocietyOrbital soft tissue surgery for patients with Treacher-Collins or Nager syndrome. A new surgical approach with early correction of soft tissue: prospective study.
The British journal of oral & maxillofacial surgeryAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Nager.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Nager
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Nager Syndrome Revisited: Integrating In Vivo and In Vitro Models to Decipher SF3B4-Dependent Tissue Coordination.
- Management and Outcomes of Neonates with Treacher Collins and Nager Syndromes.
- Transcriptome-wide profiling of alternative splicing regulators with CRISPore-seq.
- A New Case of Nager Syndrome as a Rare Cause of Acrofacial Dysostosis.
- PTBP3 Associated With 9q32 Locus Is a Candidate Gene for Nager Syndrome.
- The Development of a European Registry for Facial Dysostosis Syndromes: A Delphi-Guided Approach.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:245(Orphanet)
- OMIM OMIM:154400(OMIM)
- MONDO:0007943(MONDO)
- GARD:498(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q12811075(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome Nager
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata