Doença rara dos anexos oculares caracterizada por um fenótipo alargado da síndrome de blefarofimose, ptose, epicanto inverso e telecanto (BPES). Quando um BPES é causado por uma microdeleção que abrange outros genes além do gene causador <i>FOXL2</i>, o doente apresenta características adicionais, incluindo perturbação do desenvolvimento intelectual, anomalias genitais externas, diplegia espástica e atraso na linguagem. Também pode ser observada microcefalia adquirida.
Introdução
O que você precisa saber de cara
Visão geral
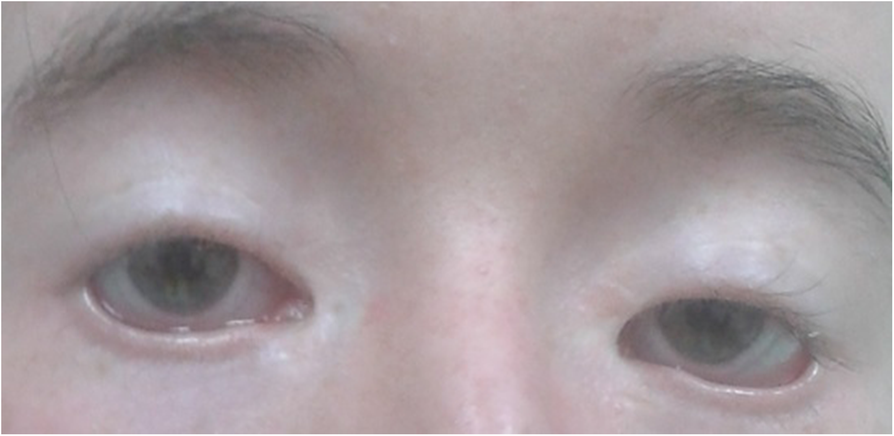
A Síndrome plus de blefarofimose-ptose-epicanto inverso é uma condição genética rara que afeta principalmente o desenvolvimento das pálpebras e, em alguns casos, a função dos ovários. O nome descreve as principais características oculares: abertura palpebral estreita (blefarofimose), queda da pálpebra superior (ptose) e uma prega cutânea que vai do canto interno do olho para baixo (epicanto inverso). O termo "plus" indica que podem estar presentes outras alterações, como problemas na glândula lacrimal e no sistema reprodutivo feminino. A condição está presente desde o nascimento (início antenatal).[1][3]
Sinais e sintomas
Os sinais e sintomas mais frequentes envolvem os olhos e as pálpebras. Além da blefarofimose, ptose e epicanto inverso, podem ocorrer: telecanto (aumento da distância entre os cantos internos dos olhos), estrabismo, nistagmo (movimentos involuntários dos olhos), ambliopia (olho preguiçoso), anormalidades da refração (miopia, hipermetropia ou astigmatismo) e ectrópio (pálpebra virada para fora). A glândula lacrimal pode estar ausente (aplasia) ou malformada, levando à diminuição da produção de lágrimas (lacrimação diminuída) e alterações nos ductos e pontos lacrimais.[1][3]
Características faciais adicionais incluem sobrancelhas muito arqueadas e espessas, ponte nasal ampla, filtro curto (espaço entre o nariz e o lábio superior) e orelhas de implantação baixa. Em mulheres, podem ocorrer alterações hormonais e reprodutivas, como hipogonadismo hipergonadotrófico (ovários que não produzem estrogênio adequadamente), ovários policísticos, hipoplasia do útero, oligomenorreia ou amenorreia secundária (ciclos menstruais irregulares ou ausentes), fertilidade diminuída e níveis baixos de estradiol sérico.[1][3]
Causas genéticas
A síndrome é causada por alterações (variantes patogênicas) no gene FOXL2 (Forkhead box protein L2). Esse gene fornece instruções para a produção de uma proteína que atua como fator de transcrição, regulando a atividade de outros genes importantes para o desenvolvimento das pálpebras e dos ovários. Mutações no FOXL2 podem levar aos sinais oculares e, em algumas mulheres, à insuficiência ovariana precoce.[1][4]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sinais oculares e faciais característicos, e pode ser confirmado por testes genéticos. O sequenciamento completo do exoma (WES) é um dos métodos disponíveis para identificar variantes no gene FOXL2. Outros exames laboratoriais que podem auxiliar no diagnóstico incluem cariótipo com bandas G, Q ou R, pesquisa de microdeleções/microduplicações por FISH e dosagem de alfa-fetoproteína. Atualmente, há 214 variantes associadas à condição registradas no ClinVar e 336 testes genéticos disponíveis.[1][4]
Tratamento e manejo
O manejo da síndrome é multidisciplinar e deve ser individualizado. Para as alterações oculares, o tratamento pode incluir correção cirúrgica da ptose e blefarofimose, uso de óculos para ambliopia e anormalidades da refração, e lubrificantes oculares para a lacrimação diminuída. O estrabismo e o nistagmo podem ser tratados com cirurgia ou terapia visual. Para as mulheres com envolvimento ovariano, o acompanhamento com endocrinologista e ginecologista é importante para avaliar a função hormonal, a fertilidade e considerar opções como reposição hormonal ou preservação da fertilidade. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para essa condição, incluindo atendimento em reabilitação para doenças raras.[1]
Prognóstico e qualidade de vida
O prognóstico depende da gravidade dos sintomas oculares e da presença de envolvimento ovariano. Com intervenções adequadas, a maioria das pessoas consegue ter boa qualidade de vida, embora possam necessitar de acompanhamento oftalmológico e endocrinológico ao longo da vida. A fertilidade pode ser afetada em mulheres, mas existem opções de planejamento familiar. Não há dados disponíveis sobre a prevalência exata da condição.[1][3]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Doença rara dos anexos oculares caracterizada por um fenótipo alargado da síndrome de blefarofimose, ptose, epicanto inverso e telecanto (BPES). Quando um BPES é causado por uma microdeleção que abrange outros genes além do gene causador <i>FOXL2</i>, o doente apresenta características adicionais, incluindo perturbação do desenvolvimento intelectual, anomalias genitais externas, diplegia espástica e atraso na linguagem. Também pode ser observada microcefalia adquirida.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Síndrome plus de blefarofimose-ptose-epicanto inverso é uma condição genética rara que afeta principalmente o desenvolvimento das pálpebras e, em alguns casos, a função dos ovários. O nome descreve as principais características oculares: abertura palpebral estreita (blefarofimose), queda da pálpebra superior (ptose) e uma prega cutânea que vai do canto interno do olho para baixo (epicanto inverso). O termo "plus" indica que podem estar presentes outras alterações, como problemas na glândula lacrimal e no sistema reprodutivo feminino. A condição está presente desde o nascimento (início antenatal).[1][3]
Sinais e sintomas
Os sinais e sintomas mais frequentes envolvem os olhos e as pálpebras. Além da blefarofimose, ptose e epicanto inverso, podem ocorrer: telecanto (aumento da distância entre os cantos internos dos olhos), estrabismo, nistagmo (movimentos involuntários dos olhos), ambliopia (olho preguiçoso), anormalidades da refração (miopia, hipermetropia ou astigmatismo) e ectrópio (pálpebra virada para fora). A glândula lacrimal pode estar ausente (aplasia) ou malformada, levando à diminuição da produção de lágrimas (lacrimação diminuída) e alterações nos ductos e pontos lacrimais.[1][3]
Características faciais adicionais incluem sobrancelhas muito arqueadas e espessas, ponte nasal ampla, filtro curto (espaço entre o nariz e o lábio superior) e orelhas de implantação baixa. Em mulheres, podem ocorrer alterações hormonais e reprodutivas, como hipogonadismo hipergonadotrófico (ovários que não produzem estrogênio adequadamente), ovários policísticos, hipoplasia do útero, oligomenorreia ou amenorreia secundária (ciclos menstruais irregulares ou ausentes), fertilidade diminuída e níveis baixos de estradiol sérico.[1][3]
Causas genéticas
A síndrome é causada por alterações (variantes patogênicas) no gene FOXL2 (Forkhead box protein L2). Esse gene fornece instruções para a produção de uma proteína que atua como fator de transcrição, regulando a atividade de outros genes importantes para o desenvolvimento das pálpebras e dos ovários. Mutações no FOXL2 podem levar aos sinais oculares e, em algumas mulheres, à insuficiência ovariana precoce.[1][4]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sinais oculares e faciais característicos, e pode ser confirmado por testes genéticos. O sequenciamento completo do exoma (WES) é um dos métodos disponíveis para identificar variantes no gene FOXL2. Outros exames laboratoriais que podem auxiliar no diagnóstico incluem cariótipo com bandas G, Q ou R, pesquisa de microdeleções/microduplicações por FISH e dosagem de alfa-fetoproteína. Atualmente, há 214 variantes associadas à condição registradas no ClinVar e 336 testes genéticos disponíveis.[1][4]
Tratamento e manejo
O manejo da síndrome é multidisciplinar e deve ser individualizado. Para as alterações oculares, o tratamento pode incluir correção cirúrgica da ptose e blefarofimose, uso de óculos para ambliopia e anormalidades da refração, e lubrificantes oculares para a lacrimação diminuída. O estrabismo e o nistagmo podem ser tratados com cirurgia ou terapia visual. Para as mulheres com envolvimento ovariano, o acompanhamento com endocrinologista e ginecologista é importante para avaliar a função hormonal, a fertilidade e considerar opções como reposição hormonal ou preservação da fertilidade. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para essa condição, incluindo atendimento em reabilitação para doenças raras.[1]
Prognóstico e qualidade de vida
O prognóstico depende da gravidade dos sintomas oculares e da presença de envolvimento ovariano. Com intervenções adequadas, a maioria das pessoas consegue ter boa qualidade de vida, embora possam necessitar de acompanhamento oftalmológico e endocrinológico ao longo da vida. A fertilidade pode ser afetada em mulheres, mas existem opções de planejamento familiar. Não há dados disponíveis sobre a prevalência exata da condição.[1][3]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 14 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 35 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição.
Transcriptional regulator. Critical factor essential for ovary differentiation and maintenance, and repression of the genetic program for somatic testis determination. Prevents trans-differentiation of ovary to testis through transcriptional repression of the Sertoli cell-promoting gene SOX9 (By similarity). Has apoptotic activity in ovarian cells. Suppresses ESR1-mediated transcription of PTGS2/COX2 stimulated by tamoxifen (By similarity). Is a regulator of CYP19 expression (By similarity). Par
Nucleus
Blepharophimosis, ptosis, and epicanthus inversus syndrome
A disorder characterized by eyelid dysplasia, small palpebral fissures, drooping eyelids and a skin fold curving in the mediolateral direction, inferior to the inner canthus. In type I BPSE (BPES1) eyelid abnormalities are associated with female infertility. Affected females show an ovarian deficit due to primary amenorrhea or to premature ovarian failure (POF). In type II BPSE (BPES2) affected individuals show only the eyelid defects.
Variantes genéticas (ClinVar)
214 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome plus de blefarofimose-ptose-epicanto inverso
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Publicações recentes
Genetic and Clinical Features of FOXL2-Associated Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome Based on 11 Chinese Families and Literature Review.
One-stage versus two-stage surgical correction of blepharophimosis-ptosis-epicanthus inversus syndrome: a retrospective comparative study.
A rare case of Blepharophimosis-Ptosis-Epicanthus inversus syndrome (BPES) associated with keratoconus: a multidisciplinary approach to diagnosis and management.
Blepharophimosis-ptosis-epicanthus inversus syndrome (BPES) due to polyAla site variant in FOXL2: diagnostic challenges with NGS.
Chromosome 3q22.2-q26.2 Interstitial Deletion in a Patient With Wisconsin Syndrome, Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome, Dandy-Walker Malformation, Pierre Robin Sequence, and Recurrent Infections.
📚 EuropePMC1 artigos no totalmostrando 1
Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome plus de blefarofimose-ptose-epicanto inverso.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome plus de blefarofimose-ptose-epicanto inverso
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- "Blepharophimosis-plus" syndromes: Frequency of systemic genetic disorders that also include blepharophimosis.
- Genetic and Clinical Features of FOXL2-Associated Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome Based on 11 Chinese Families and Literature Review.
- One-stage versus two-stage surgical correction of blepharophimosis-ptosis-epicanthus inversus syndrome: a retrospective comparative study.
- A rare case of Blepharophimosis-Ptosis-Epicanthus inversus syndrome (BPES) associated with keratoconus: a multidisciplinary approach to diagnosis and management.
- Blepharophimosis-ptosis-epicanthus inversus syndrome (BPES) due to polyAla site variant in FOXL2: diagnostic challenges with NGS.
- Chromosome 3q22.2-q26.2 Interstitial Deletion in a Patient With Wisconsin Syndrome, Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome, Dandy-Walker Malformation, Pierre Robin Sequence, and Recurrent Infections.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:572333(Orphanet)
- MONDO:0035521(MONDO)
- GARD:22312(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome plus de blefarofimose-ptose-epicanto inverso
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)