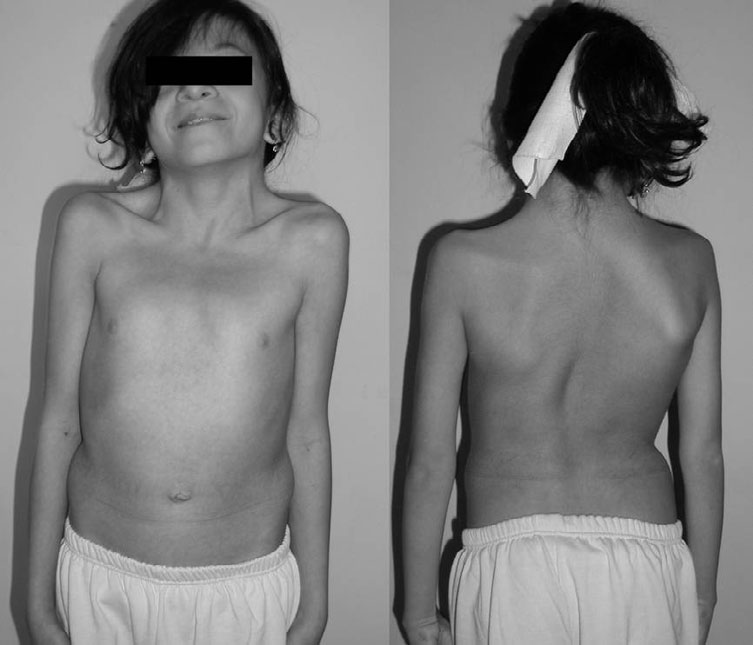
Acredita-se que a síndrome de Watson seja uma variante da neurofibromatose tipo 1. Os sintomas dessa condição são estenose valvar pulmonar, manchas café com leite e baixa estatura. As pontuações do teste de QI para indivíduos com síndrome de Watson podem variar entre 60-100. Muitas pessoas com essa condição também apresentam tamanho de cabeça maior que a média (macrocefalia) e nódulos de Lisch. Embora mutações no gene NF1 tenham sido encontradas em famílias com síndrome de Watson, a causa exata desta condição é desconhecida. A condição é herdada em um padrão autossômico dominante. O tratamento visa controlar os sintomas específicos de um indivíduo.
Introdução
O que você precisa saber de cara
Acredita-se que a síndrome de Watson seja uma variante da neurofibromatose tipo 1. Os sintomas dessa condição são estenose valvar pulmonar, manchas café com leite e baixa estatura. As pontuações do teste de QI para indivíduos com síndrome de Watson podem variar entre 60-100. Muitas pessoas com essa condição também apresentam tamanho de cabeça maior que a média (macrocefalia) e nódulos de Lisch. Embora mutações no gene NF1 tenham sido encontradas em famílias com síndrome de Watson, a causa exata desta condição é desconhecida. A condição é herdada em um padrão autossômico dominante. O tratamento visa controlar os sintomas específicos de um indivíduo.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 7 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 16 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição.
Stimulates the GTPase activity of Ras. NF1 shows greater affinity for Ras GAP, but lower specific activity. May be a regulator of Ras activity
NucleusNucleus, nucleolusCell membrane
Neurofibromatosis 1
A disease characterized by patches of skin pigmentation (cafe-au-lait spots), Lisch nodules of the iris, tumors in the peripheral nervous system and fibromatous skin tumors. Individuals with the disorder have increased susceptibility to the development of benign and malignant tumors.
Variantes genéticas (ClinVar)
7.190 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Watson
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Speech and language skills in a case of Watson syndrome.
Watson syndrome is a rare genetic condition partly characterised by developmental delays and learning difficulties. A profile of speech and language skills associated with this developmental syndrome is yet to be described in the literature. In order to address this gap, this study presents the case of an 18-year-old man with Watson syndrome and reports both standardised and naturalistic assessments of speech, language, oro-motor skills, and semantic and phonemic fluency. Analyses included norm-referencing, discrepancy comparison, phonological process analysis, and acoustic analyses of voice and conversational fluency. The participant's semantic fluency approximated to the 84th percentile and the vocabulary, voice, and receptive language measures were within standard normative range. In contrast, expressive language difficulties and articulatory difficulties associated with impaired oro-motor skills were apparent. Specific tongue-motor difficulty impeded oral diadochokinesis, with gliding, stopping, and cluster reduction among the phonological processes observed to mitigate oro-motor difficulties. Language scores were lowest on tasks of working memory, syntax, and pragmatics, however neither syntax nor pragmatics presented increased difficulty in naturalistic conversation, indicating an influence of reduced working memory on language performance. The absence of explicit cognitive or communicative difficulty suggests specific speech and language difficulties. To conclude, the findings are discussed from both clinical and theoretical perspectives. Watson syndrome and the aetiology of communication difficulties are suggested as directions for future research, to validate these findings and diversify understanding of inclusive communication approaches.
A rare case of Watson syndrome.
Watson for the first time reported a case series of children in a family that presented with pulmonary valve stenosis, mental retardation, short stature, and small brown color skin lesions that are known as cafe-au-lait spots. We present a rare new variant of the syndrome in an adult patient with severe pulmonary valve stenosis, main, left, and right pulmonary artery aneurysm, short stature, mental retardation, coronary artery disease, and skin lesions. The patient underwent open cardiac surgery with pulmonary valvotomy and aneurysmorrhaphy of the main pulmonary artery and its right and left branches. The postoperative course was uneventful and the six-month follow-up with transthoracic echocardiography revealed no recurrence of aneurysm of repairing pulmonary arteries and good clinical outcome of the patient. Our patient had a unique characteristic of aneurysm of the main pulmonary artery and its both branches that has rarely been reported previously in the medical literature.
Expanding the Noonan spectrum/RASopathy NGS panel: Benefits of adding NF1 and SPRED1.
RASopathies are a group of disorders caused by disruptions to the RAS-MAPK pathway. Despite being in the same pathway, Neurofibromatosis Type 1 (NF1) and Legius syndrome (LS) typically present with phenotypes distinct from Noonan spectrum disorders (NSDs). However, some NF1/LS individuals also exhibit NSD phenotypes, often referred to as Neurofibromatosis-Noonan syndrome (NFNS), and may be mistakenly evaluated for NSDs, delaying diagnosis, and affecting patient management. A derivation cohort of 28 patients with a prior negative NSD panel and either NFNS or a suspicion of NSD and café-au-lait spots underwent NF1 and SPRED1 sequencing. To further determine the utility and burden of adding these genes, a validation cohort of 505 patients with a suspected RASopathy were tested on a 14-gene RASopathy-associated panel. In the derivation cohort, six (21%) patients had disease-causing NF1 or SPRED1 variants. In the validation cohort, 11 (2%) patients had disease-causing variants and 15 (3%) had variants of uncertain significance in NF1 or SPRED1. Of those with disease-causing variants, 5/17 only had an NSD diagnosis. Adding NF1 and SPRED1 to RASopathy panels can speed diagnosis and improve patient management, without significantly increasing the burden of inconclusive results.
Neurofibromin 1 Impairs Natural Killer T-Cell-Dependent Antitumor Immunity against a T-Cell Lymphoma.
Neurofibromin 1 (NF1) is a tumor suppressor gene encoding a Ras GTPase that negatively regulates Ras signaling pathways. Mutations in NF1 are linked to neurofibromatosis type 1, juvenile myelomonocytic leukemia and Watson syndrome. In terms of antitumor immunity, CD1d-dependent natural killer T (NKT) cells play an important role in the innate antitumor immune response. Generally, Type-I NKT cells protect (and Type-II NKT cells impair) host antitumor immunity. We have previously shown that CD1d-mediated antigen presentation to NKT cells is regulated by cell signaling pathways. To study whether a haploinsufficiency in NF1 would affect CD1d-dependent activation of NKT cells, we analyzed the NKT-cell population as well as the functional expression of CD1d in Nf1+/- mice. Nf1+/- mice were found to have similar levels of NKT cells as wildtype (WT) littermates. Interestingly, however, reduced CD1d expression was observed in Nf1+/- mice compared with their WT littermates. When inoculated with a T-cell lymphoma in vivo, Nf1+/- mice survived longer than their WT littermates. Furthermore, blocking CD1d in vivo significantly enhanced antitumor activity in WT, but not in Nf1+/- mice. In contrast, a deficiency in Type-I NKT cells increased antitumor activity in Nf1+/- mice, but not in WT littermates. Therefore, these data suggest that normal NF1 expression impairs CD1d-mediated NKT-cell activation and antitumor activity against a T-cell lymphoma.
Generation of KCL025 research grade human embryonic stem cell line carrying a mutation in NF1 gene.
The KCL025 human embryonic stem cell line was derived from an embryo donated for research that carried an autosomal dominant mutation in the NF1 gene encoding neurofibromin (c.3739-3742 ΔTTTG). Mutations in this gene have been linked to neurofibromatosis type 1, juvenile myelomonocytic leukemia and Watson syndrome. The ICM was isolated using laser microsurgery and plated on γ-irradiated human foreskin fibroblasts. Both the derivation and cell line propagation were performed in an animal product-free environment. Pluripotent state and differentiation potential were confirmed by in vitro assays.
Publicações recentes
Speech and language skills in a case of Watson syndrome.
A rare case of Watson syndrome.
Expanding the Noonan spectrum/RASopathy NGS panel: Benefits of adding NF1 and SPRED1.
Neurofibromin 1 Impairs Natural Killer T-Cell-Dependent Antitumor Immunity against a T-Cell Lymphoma.
Generation of KCL025 research grade human embryonic stem cell line carrying a mutation in NF1 gene.
📚 EuropePMC10 artigos no totalmostrando 6
Speech and language skills in a case of Watson syndrome.
Clinical linguistics & phoneticsA rare case of Watson syndrome.
Folia medicaExpanding the Noonan spectrum/RASopathy NGS panel: Benefits of adding NF1 and SPRED1.
Molecular genetics & genomic medicineNeurofibromin 1 Impairs Natural Killer T-Cell-Dependent Antitumor Immunity against a T-Cell Lymphoma.
Frontiers in immunologyGeneration of KCL025 research grade human embryonic stem cell line carrying a mutation in NF1 gene.
Stem cell researchGeneration of KCL024 research grade human embryonic stem cell line carrying a mutation in NF1 gene.
Stem cell researchAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Watson.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Watson
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Speech and language skills in a case of Watson syndrome.
- A rare case of Watson syndrome.
- Expanding the Noonan spectrum/RASopathy NGS panel: Benefits of adding NF1 and SPRED1.
- Neurofibromin 1 Impairs Natural Killer T-Cell-Dependent Antitumor Immunity against a T-Cell Lymphoma.
- Generation of KCL025 research grade human embryonic stem cell line carrying a mutation in NF1 gene.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:3444(Orphanet)
- OMIM OMIM:193520(OMIM)
- MONDO:0008672(MONDO)
- GARD:5540(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q1112752(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome Watson
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata