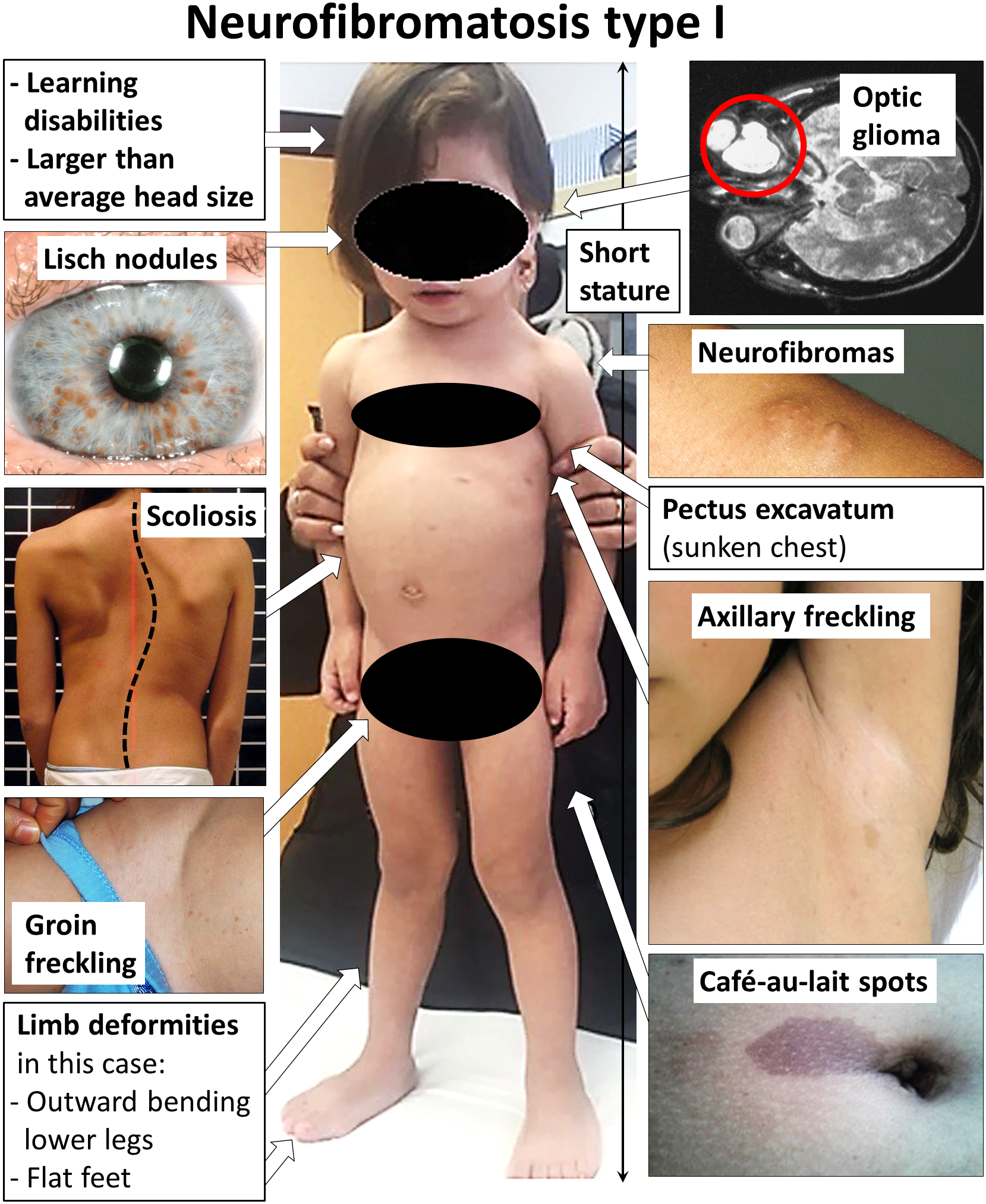
Uma forma rara e grave de neurofibromatose tipo 1 (NF1), caracterizada por características faciais um pouco diferentes, atraso no desenvolvimento, deficiência intelectual, maior risco de câncer e um grande número de neurofibromas.
Introdução
O que você precisa saber de cara
Uma forma rara e grave de neurofibromatose tipo 1 (NF1), caracterizada por características faciais um pouco diferentes, atraso no desenvolvimento, deficiência intelectual, maior risco de câncer e um grande número de neurofibromas.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 50 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 137 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Not applicable.
Stimulates the GTPase activity of Ras. NF1 shows greater affinity for Ras GAP, but lower specific activity. May be a regulator of Ras activity
NucleusNucleus, nucleolusCell membrane
Neurofibromatosis 1
A disease characterized by patches of skin pigmentation (cafe-au-lait spots), Lisch nodules of the iris, tumors in the peripheral nervous system and fibromatous skin tumors. Individuals with the disorder have increased susceptibility to the development of benign and malignant tumors.
E2-dependent E3 ubiquitin-protein ligase that functions as a RIGI coreceptor in the sensing of viral RNAs in cell cytoplasm and the activation of the antiviral innate immune response (PubMed:19017631, PubMed:19484123, PubMed:21147464, PubMed:23950712, PubMed:28469175, PubMed:31006531). Together with the UBE2D3, UBE2N and UB2V1 E2 ligases, catalyzes the 'Lys-63'-linked polyubiquitination of RIGI oligomerized on viral RNAs, an essential step in the activation of the RIG-I signaling pathway (PubMed
CytoplasmCytoplasm, Stress granule
Variantes genéticas (ClinVar)
7.251 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
10 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de microdeleção 17q11
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Genetic/epigenetic effects in NF1 microdeletion syndrome: beyond the haploinsufficiency, looking at the contribution of not deleted genes.
NF1 microdeletion syndrome, accounting for 5-11% of NF1 patients, is caused by a deletion in the NF1 region and it is generally characterized by a severe phenotype. Although 70% of NF1 microdeletion patients presents the same 1.4 Mb type-I deletion, some patients may show additional clinical features. Therefore, the contribution of several pathogenic mechanisms, besides haploinsufficiency of some genes within the deletion interval, is expected and needs to be defined. We investigated an altered expression of deletion flanking genes by qPCR in patients with type-1 NF1 deletion, compared to healthy donors, possibly contributing to the clinical traits of NF1 microdeletion syndrome. In addition, the 1.4-Mb deletion leads to changes in the 3D chromatin structure in the 17q11.2 region. Specifically, this deletion alters DNA-DNA interactions in the regions flanking the breakpoints, as demonstrated by our 4C-seq analysis. This alteration likely causes position effect on the expression of deletion flanking genes.Interestingly, 4C-seq analysis revealed that in microdeletion patients, an interaction was established between the RHOT1 promoter and the SLC6A4 gene, which showed increased expression. We performed NGS on putative modifier genes, and identified two "likely pathogenic" rare variants in RAS pathway, possibly contributing to incidental phenotypic features.This study provides new insights into understanding the pathogenesis of NF1 microdeletion syndrome and suggests a novel pathomechanism that contributes to the expression phenotype in addition to haploinsufficiency of genes located within the deletion.This is a pivotal approach that can be applied to unravel microdeletion syndromes, improving precision medicine, prognosis and patients' follow-up.
Deregulated expression of polycomb repressive complex 2 target genes in a NF1 patient with microdeletion generating the RNF135-SUZ12 chimeric gene.
Neurofibromatosis type I (NF1) microdeletion syndrome, accounting for 5-11% of NF1 patients, is caused by the heterozygous deletion of NF1 and a variable number of flanking genes in the 17q11.2 region. This syndrome is characterized by more severe symptoms than those shown by patients with intragenic NF1 mutation and by variable expressivity, which is not fully explained by the haploinsufficiency of the genes included in the deletions. We here reevaluate an 8-year-old NF1 patient, who carries an atypical deletion generating the RNF135-SUZ12 chimeric gene, previously described when he was 3 years old. As the patient has developed multiple cutaneous/subcutaneous neurofibromas over the past 5 years, we hypothesized a role of RNF135-SUZ12 chimeric gene in the onset of the patient's tumor phenotype. Interestingly, SUZ12 is generally lost or disrupted in NF1 microdeletion syndrome and frequently associated to cancer as RNF135. Expression analysis confirmed the presence of the chimeric gene transcript and revealed hypo-expression of five out of the seven analyzed target genes of the polycomb repressive complex 2 (PRC2), to which SUZ12 belongs, in the patient's peripheral blood, indicating a higher transcriptional repression activity mediated by PRC2. Furthermore, decreased expression of tumor suppressor gene TP53, which is targeted by RNF135, was detected. These results suggest that RNF135-SUZ12 chimera may acquire a gain of function, compared with SUZ12 wild type in the PRC2 complex, and a loss of function relative to RNF135 wild type. Both events may have a role in the early onset of the patient's neurofibromas.
Concomitance of 47,XXY, a balanced reciprocal translocation of t(4;17)(q12;q11.2) encompassing SPINK2 at 4q12 and NOS at 17q11.2 and an AZFa sY86 deletion in an infertile male.
We present an infertile male who was incidentally detected to have Klinefelter syndrome, a balanced reciprocal translocation of t(4; 17) (q12; q11.2) and an AZFa sY86 deletion. We review the literature and discuss the significance of 47,XXY, t(4; 17) (q12; q11.2) and AZFa sY86 deletion in this case. A 37-year-old married infertile male was referred for genetic studies of azoospermia. His height was 195 cm and his weight was 85 kg. He had been married for more than one year without any pregnancy in his wife. He was referred for genetic counseling. Cytogenetic analysis revealed a karyotype of 47,XXY,t(4; 17) (q12; q11.2). In addition to Klinefelter syndrome, a balanced reciprocal translocation and an AZFa microdeletion were found. Sequence analysis of SPINK2 and NOS was also performed. These two fertile related genes were located at the breakpoints of translocation respectively. Heterozygosity of single-nucleotide polymorphisms (SNPs) evidenced the presence of two alleles as well as no deletions occurred at the breakpoint regions. An AZF gene analysis revealed a microdeletion at the region of AZFa sY86 region. Genetic analysis of an infertile male may detect multiple factors associated with azoospermia such as translocation, an AZF deletion and Klinefelter syndrome. This case emphasized the importance of tests for chromosomes and AZF deletions among patients with azoospermia. Complete genetic counseling of the consequence of a familial inheritance is also necessary to detect more family carrier members for the prevention of unbalanced chromosome in the offspring.
[Neurofibromatosis-1 microdeletion syndrome.].
Neurofibromatosis type 1 is a clinically extremely heterogeneous neurocutaneous disorder, inherited in autosomal dominant manner. It is primarily caused by intragenic loss-of-function mutations in the NF1 gene, however, as a result of improvements in molecular diagnostics, copy number variants affecting the NF1 gene and its flanking regions are increasingly being detected. Based on genotype-phenotype analyses, two groups can be distinguished: neurofibromatosis type 1 caused by point mutations and the so-called 17q11.2 microdeletion syndrome caused by microdeletions. Microdeletions are observed in 5-10% of cases and can be divided into four different types (type 1, 2, 3 and atypical) according to the size of the deletion, the genomic location of the breakpoints and the affected gene content. Patients with microdeletions often have a more severe course of the disease, with an increased risk of malignancies. With this review, which summarizes the main characteristics and molecular genetic background of neurofibromatosis-1 microdeletion syndrome, we would like to emphasize the importance of early diagnosis of patients with microdeletion syndrome and draw attention to the importance of close follow-up. Orv Hetil. 2022; 163(51): 2041-2051. Az 1-es típusú neurofibromatosis autoszomális domináns öröklésmenetet mutató, klinikailag rendkívül heterogén neurocutan kórkép, amelynek kialakulásában elsődlegesen az NF1-gén intragenikus funkcióvesztéses mutációi játszanak szerepet. Ugyanakkor a molekuláris diagnosztika fejlődésének köszönhetően egyre több esetben sikerül kimutatni az NF1-gént és az azzal szomszédos régiókat érintő kópiaszámbeli variánsokat. Genotípus-fenotípus elemzések alapján a pontmutációs eltérések okozta 1-es típusú neurofibromatosis, illetve a microdeletiós eltérések okozta, ún. 17q11.2 microdeletiós szindróma elkülöníthetők egymástól. Microdeletiók az esetek 5–10%-ában figyelhetők meg, melyek méretük, töréspontjaik genomi lokalizációja és érintett géntartalmuk alapján négy különböző típusba (1-es, 2-es, 3-as és atípusos) sorolhatók. A microdeletiós betegek gyakran súlyosabb kórlefolyást mutatnak, melyből kiemelendő a malignitások emelkedett kockázata. Az összefoglaló közleménnyel, mely a neurofibromatosis-1 microdeletiós szindróma főbb jellemzőit, molekuláris genetikai hátterét és vizsgálati módszereit tárgyalja, a microdeletiós szindrómás betegek korai diagnózishoz jutásának fontosságát szeretnénk hangsúlyozni és felhívni a figyelmet a szoros nyomon követés jelentőségére. Orv Hetil. 2022; 163(51): 2041–2051.
Co-existing bipolar disease and 17q12 deletion: a rare case report.
17q12 microdeletion syndrome is a rare autosomal dominant chromosomal anomaly, caused by the deletion of a 1.4 Mb-spanning DNA sequence on the long arm of chromosome 17. Herein, we report the first bipolar disease (BPD) case with a 1.6-Mb deletion in the 17q11.2-17q12 chromosome region. Physical examination of the case was performed. Karyotype and microarray analyses were performed for the case and the parents. Physical examination revealed mild dysmorphic features such as high and forehead, full cheeks, slightly depressed nasal bridge and arched eyebrow. Chromosomal analysis of the patient revealed 46, XX, del(17)(q12) karyotype, and parents' karyotype were normal. In the microarray analysis of patient, 1.6 megabases deletion was detected in the 17q12 region [arr(hg19) 17q12 (34,611,352-36,248,918) ×1]. The microarray analysis of the mother was normal. The father's microarray showed 473 kilobases duplication in the 11p11.12 region. Although 17q12 deletion syndrome has been associated with bipolar disorder, very few such cases have been described in the literature. Genetic counseling should be considered in patients with remarkable phenotype, complex symptomatology, neurodevelopmental disorder and additional conspicuous medical conditions.
Publicações recentes
SPAG7 is a candidate gene for the periodic fever, aphthous stomatitis, pharyngitis and adenopathy (PFAPA) syndrome.
Positive regulation of apoptosis by HCA66, a new Apaf-1 interacting protein, and its putative role in the physiopathology of NF1 microdeletion syndrome patients.
NF1 microdeletion syndrome: refined FISH characterization of sporadic and familial deletions with locus-specific probes.
📚 EuropePMCmostrando 18
Genetic/epigenetic effects in NF1 microdeletion syndrome: beyond the haploinsufficiency, looking at the contribution of not deleted genes.
Human geneticsDeregulated expression of polycomb repressive complex 2 target genes in a NF1 patient with microdeletion generating the RNF135-SUZ12 chimeric gene.
NeurogeneticsConcomitance of 47,XXY, a balanced reciprocal translocation of t(4;17)(q12;q11.2) encompassing SPINK2 at 4q12 and NOS at 17q11.2 and an AZFa sY86 deletion in an infertile male.
Taiwanese journal of obstetrics & gynecology[Neurofibromatosis-1 microdeletion syndrome.].
Orvosi hetilapCo-existing bipolar disease and 17q12 deletion: a rare case report.
Psychiatric geneticsPatient-derived iPSC-cerebral organoid modeling of the 17q11.2 microdeletion syndrome establishes CRLF3 as a critical regulator of neurogenesis.
Cell reportsGenotype-Phenotype Associations in Patients With Type-1, Type-2, and Atypical NF1 Microdeletions.
Frontiers in geneticsA novel MEIS2 mutation explains the complex phenotype in a boy with a typical NF1 microdeletion syndrome.
European journal of medical genetics[A genetic case study of neurofibromatosis type 1-microdeletion syndrome caused by atypical 17q11.2 microdeletion].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsNF1 microdeletion syndrome: case report of two new patients.
Italian journal of pediatricsRare NF1 microdeletion syndrome in an Omani patient.
Clinical case reportsAn estimation of the prevalence of genomic disorders using chromosomal microarray data.
Journal of human geneticsInterstitial microdeletion of 17q11.2 is associated with hypotonia, fatigue, intellectual disability, and a subtle facial phenotype in three unrelated patients.
American journal of medical genetics. Part A[Phenotypic and genetic analysis of a child carrying a 17q11.2 microdeletion].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsIdentification of an atypical microdeletion generating the RNF135-SUZ12 chimeric gene and causing a position effect in an NF1 patient with overgrowth.
Human geneticsCharacterization of the Phenotype Associated with Microduplication Reciprocal to NF1 Microdeletion Syndrome.
Cytogenetic and genome researchA novel de novo microdeletion at 17q11.2 adjacent to NF1 gene associated with developmental delay, short stature, microcephaly and dysmorphic features.
Molecular cytogenetics[Prenatal genetic diagnosis for a fetus with atypical neurofibromatosis type 1 microdeletion].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de microdeleção 17q11.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de microdeleção 17q11
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Genetic/epigenetic effects in NF1 microdeletion syndrome: beyond the haploinsufficiency, looking at the contribution of not deleted genes.
- Deregulated expression of polycomb repressive complex 2 target genes in a NF1 patient with microdeletion generating the RNF135-SUZ12 chimeric gene.
- Concomitance of 47,XXY, a balanced reciprocal translocation of t(4;17)(q12;q11.2) encompassing SPINK2 at 4q12 and NOS at 17q11.2 and an AZFa sY86 deletion in an infertile male.
- [Neurofibromatosis-1 microdeletion syndrome.].
- Co-existing bipolar disease and 17q12 deletion: a rare case report.
- SPAG7 is a candidate gene for the periodic fever, aphthous stomatitis, pharyngitis and adenopathy (PFAPA) syndrome.
- Positive regulation of apoptosis by HCA66, a new Apaf-1 interacting protein, and its putative role in the physiopathology of NF1 microdeletion syndrome patients.
- NF1 microdeletion syndrome: refined FISH characterization of sporadic and familial deletions with locus-specific probes.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:97685(Orphanet)
- OMIM OMIM:613675(OMIM)
- MONDO:0013357(MONDO)
- GARD:5408(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q21154063(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome de microdeleção 17q11
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata