RASopatia e uma variante da neurofibromatose tipo 1 (NF1) caracterizada pela combinação de características da NF1, como manchas café com leite, nódulos de Lisch na íris, sardas axilares e inguinais, glioma do nervo óptico e neurofibromas múltiplos; e síndrome de Noonan (SN), como baixa estatura, características faciais típicas (hipertelorismo, ptose, fissuras palpebrais inclinadas para baixo, orelhas de implantação baixa e giradas posteriormente com hélice espessada e testa larga), defeitos cardíacos congênitos e deformidade pectus incomum. Como essas três entidades têm sobreposição fenotípica significativa, o teste genético molecular é frequentemente necessário para um diagnóstico correto (como quando manchas cafC)-com leite estão presentes em pacientes com diagnóstico de SN).
Introdução
O que você precisa saber de cara
RASopatia e uma variante da neurofibromatose tipo 1 (NF1) caracterizada pela combinação de características da NF1, como manchas café com leite, nódulos de Lisch na íris, sardas axilares e inguinais, glioma do nervo óptico e neurofibromas múltiplos; e síndrome de Noonan (SN), como baixa estatura, características faciais típicas (hipertelorismo, ptose, fissuras palpebrais inclinadas para baixo, orelhas de implantação baixa e giradas posteriormente com hélice espessada e testa larga), defeitos cardíacos congênitos e deformidade pectus incomum. Como essas três entidades têm sobreposição fenotípica significativa, o teste genético molecular é frequentemente necessário para um diagnóstico correto (como quando manchas cafC)-com leite estão presentes em pacientes com diagnóstico de SN).
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 16 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 49 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisCatalyzes the concomitant phosphorylation of a threonine and a tyrosine residue in a Thr-Glu-Tyr sequence located in MAP kinases. Activates the ERK1 and ERK2 MAP kinases (By similarity). Activates BRAF in a KSR1 or KSR2-dependent manner; by binding to KSR1 or KSR2 releases the inhibitory intramolecular interaction between KSR1 or KSR2 protein kinase and N-terminal domains which promotes KSR1 or KSR2-BRAF dimerization and BRAF activation (PubMed:29433126)
CytoplasmMembrane
Cardiofaciocutaneous syndrome 4
A form of cardiofaciocutaneous syndrome, a multiple congenital anomaly disorder characterized by a distinctive facial appearance, heart defects and intellectual disability. Heart defects include pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Some affected individuals present with ectodermal abnormalities such as sparse, friable hair, hyperkeratotic skin lesions and a generalized ichthyosis-like condition. Typical facial features are similar to Noonan syndrome. They include high forehead with bitemporal constriction, hypoplastic supraorbital ridges, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly angulated ears with prominent helices.
Stimulates the GTPase activity of Ras. NF1 shows greater affinity for Ras GAP, but lower specific activity. May be a regulator of Ras activity
NucleusNucleus, nucleolusCell membrane
Neurofibromatosis 1
A disease characterized by patches of skin pigmentation (cafe-au-lait spots), Lisch nodules of the iris, tumors in the peripheral nervous system and fibromatous skin tumors. Individuals with the disorder have increased susceptibility to the development of benign and malignant tumors.
Variantes genéticas (ClinVar)
7.385 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 704 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
16 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de neurofibromatose-Noonan
Centros de Referência SUS
24 centros habilitados pelo SUS para Síndrome de neurofibromatose-Noonan
Centros para Síndrome de neurofibromatose-Noonan
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
2 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
Novel Variants in PTPN11, NF1, RASA2, and MAP2K1: Expanding the Molecular Spectrum of RASopathies in a Turkish Cohort.
RASopathies are a group of genetically heterogeneous developmental disorders caused by germline variants affecting the RAS/MAPK signaling pathway. These disorders display overlapping clinical features and diverse molecular mechanisms. This study aimed to evaluate the clinical and molecular spectrum of patients diagnosed with RASopathies, with a particular focus on novel and rare variants. A retrospective, multicenter study was conducted on patients with clinically suspected RASopathy and diagnosed by targeted Next Generation Sequencing analysis between 2021 and 2024. Variants were classified according to ACMG criteria, and clinical data were reviewed for genotype-phenotype correlations. Among 23 patients (14 males, 9 females), 15 (65.2%) had Noonan syndrome, five (21.7%) Neurofibromatosis Type 1, two (8.7%) Cardio-facio-cutaneous syndrome, and one (4.3%) Neurofibromatosis-Noonan syndrome. The most frequent clinical findings were craniofacial dysmorphism (91.3%), musculoskeletal anomalies (82.6%), and cutaneous features (78.3%). A total of 24 heterozygous variants were identified in seven genes: PTPN11 (45.8%), NF1 (25%), LZTR1 (12.5%), and RASA2, SOS1, MAP2K1, and BRAF (each 4.2%). Four novel variants were detected (PTPN11 c.853 + 4A>G, RASA2 p.E71D, NF1 p.W784Mfs*10, MAP2K1 p.A106T). This study highlights the clinical and molecular heterogeneity of RASopathies and expands the variant spectrum with novel and rare pathogenic alterations. The identification of new variants, particularly in rarely implicated genes such as RASA2, underlines the diagnostic value of comprehensive NGS-based testing and the need for individualized, multidisciplinary clinical management.
Neurofibromatosis-Noonan syndrome: a prospective monocentric study of 26 patients and literature review.
Data on clinical manifestations of neurofibromatosis-Noonan syndrome (NF-NS) remain heterogeneous, with limited validated descriptions. This study aims to better define the clinical and molecular features of NF-NS and compare them with existing literature. Secondary objectives include evaluating inter-rater diagnostic agreement among experienced clinicians and assessing the utility of deep-learning algorithms (Face2Gene® [F2G]). Additionally, we assess the prevalence of congenital heart malformations (CHM) in NF-NS compared to 'classic' neurofibromatosis type 1 (NF1). A 9-year, prospective, monocentric study was conducted, involving patients with NF1 pathogenic variants (PVs) and Noonan syndrome-like facial phenotype (NSLFP). Twenty-six patients were enrolled. NSLFP was categorized as 'suggestive' in 69% of cases and 'typical' in 31%. The presence of at least two facial abnormalities (e.g., low-set ears, downslanted palpebral fissures, hypertelorism, and ptosis) was consistently observed in 'typical' cases. Inter-rater concordance was substantial (0.65 [95% CI = 0.56; 0.74]), while concordance between clinicians and F2G was almost perfect at (0.821 [CI 95% = 0.625; 1.000]). Missense NF1 PVs were observed in 38.5% of cases. Apart from NSLP and a high frequency of pectus excavatum (62.5%), no significant differences in anthropometric, dermatological, neurological, skeletal, or ocular clinical features were observed between NF-NS and 'classic' NF1. CHM were found in 19.2% of NF-NS patients, with pulmonic stenosis present in 7.7%. NF-NS is a distinct phenotypic variant of NF1, marked by NSLP with consistent facial features -, and frequent pectus excavatum. F2G demonstrated high diagnostic concordance, reinforcing its clinical utility. Given the elevated risk of CHM, especially pulmonic stenosis, proactive cardiovascular assessment similar to other RASopathies is recommended for NS-NF patients, regardless of NF1 PV type.
Molecular and Clinical Profiles of Patients with RASopathies: Targeted Next-Generation Sequencing Panel Results and Identification of 14 Novel Disease-Causing Variants.
RASopathies are among the most prevalent genetic syndromes caused by variants in the Ras/MAPK signaling pathway, affecting various systems such as the heart, craniofacial features, skin, musculoskeletal system, hearing, and vision. They can also increase the risk of secondary malignancies. Despite clinical overlaps, distinguishing features are crucial for diagnosis, as different variants lead to distinct clinical implications. This study reviews the molecular and clinical characteristics of RASopathies, focusing on neurofibromatosis type 1 (NF1) and non-NF1 RASopathies. The study analyzed 76 patients referred to our outpatient clinic over a 6-year period, all of whom were clinically diagnosed with RASopathy and confirmed in most cases by molecular testing. Patient files, clinical photographs, and laboratory results were reviewed and analyzed. A targeted multigene next-generation sequencing panel test was performed, followed by Sanger sequencing for both confirmation and segregation analysis. Multiplex ligation-dependent probe amplification was conducted in a patient with normal sequence results but strong clinical suspicion, to identify potential deletions. We identified 44 pathogenic, 25 likely pathogenic variants, and 6 variants of uncertain significance based on American College of Medical Genetics and Genomics (ACMG) criteria. Among these, 14 novel variants were found - 13 in the NF1 gene and one in SOS1. NF1 variants were present in 51 cases. Additional variants, likely to represent clinically significant findings, were identified in PTPN11 (n = 11), RAF1 (n = 4), SOS1 (n = 3), RIT1 (n = 3), KRAS (n = 1), NRAS (n = 1), SOS2 (n = 1), and BRAF (n = 1). Diagnoses included 49 patients with NF1, 21 with Noonan syndrome, 2 with neurofibromatosis-Noonan syndrome, 2 with Noonan syndrome with multiple lentigines, and 1 with cardiofaciocutaneous syndrome. Here, 12% of NF1 variants were located in exon 21, 36% of PTPN11 variants in exon 3, and 75% of RAF1 variants in exon 7. RASopathies have a broad molecular and clinical spectrum, complicating diagnosis and management. Accurate clinical correlation and molecular analysis are essential, as different RASopathy syndromes can result from variants in the same genes, while the same syndrome may arise from different genetic alterations. This study identifies novel variants and emphasizes the need for precise diagnostic approaches in these complex disorders.
Clinical and Genetic Characterization of Noonan Syndrome in a Romanian Cohort from Transylvania: Details on PTPN11 c.922A>G Variant and Phenotypic Spectrum.
Background: Noonan syndrome (NS) is a genetically heterogeneous condition within the RASopathies spectrum, with distinctive craniofacial features, congenital heart defects, short stature, and variably present developmental delay. Most cases result from variants in genes regulating the RAS/MAPK pathway, with PTPN11 variants being the most frequent; the c.922A>G substitution being among the most commonly reported. Methods: This pilot study analyzed clinical and partial genetic features of NS in a cohort from Transylvania, evaluated in the Children's Emergency Clinical Hospital in Cluj-Napoca. Thirty-one patients fulfilling the Van der Burgt diagnostic criteria (twenty-two males, nine females) were included. Clinical data were systematically reviewed, and targeted molecular testing for the PTPN11 c.922A>G variant was performed. Results: Congenital heart defects were highly prevalent, with pulmonary stenosis representing the most frequent anomaly (54.8%). Craniofacial dysmorphism was observed in 76.7% of cases, cryptorchidism in 50% of the males, and short stature below the third percentile was described in 77.4% of patients. Genetic screening identified the PTPN11 c.922A>G variant in two individuals (6.45%). Additional diagnoses included Williams-Beuren syndrome and a 17q11.2 deletion consistent with Neurofibromatosis-Noonan syndrome, underscoring the clinical and genetic heterogeneity of the cohort. Comparison with international reports highlighted variability in phenotype and variant frequency. Future research directions include Sanger sequencing of key PTPN11 exons and the application of next-generation sequencing targeting all RAS pathway genes. Conclusions: This is the first Romanian cohort study on patients with a clinical suspicion of NS, providing insight into their evaluation. The findings reinforce the need for comprehensive molecular approaches, facilitating diagnostic precision and counseling strategies.
Expanding the Genetic Landscape of RASopathies: Significance of Including NF1 in Targeted Panels.
RASopathies are genetic disorders linked to the RAS/MAPK signaling pathway, including Noonan syndrome and related conditions. These disorders have overlapping features and variable phenotypes, making diagnosis challenging. This study examines the clinical and genetic characteristics of RASopathies in pediatric patients to improve diagnostic accuracy and identify novel pathogenic variants. A total of 23 patients, who were not related to each other and were clinically diagnosed with RASopathy, participated in the study. Patients underwent next-generation sequencing (NGS) panel analyses of 14 RASopathy genes. Pathogenic variants were found in 18 out of 23 patients (78%). The most common variants were in PTPN11 and SOS1. Novel variants were identified in KRAS and NF1 expanding the known genetic spectrum. Frequent clinical features included distinctive facial features, growth delays, and heart defects, notably pulmonary stenosis. Intellectual disability was more common in patients with PTPN11 variants, while SOS1 variants were associated with unique features such as previously unreported polydactyly and choanal atresia. Targeted NGS panels improve diagnosis of RASopathies, with a variant detection rate of 78%. Including the NF1 gene, even without signs of neurofibromatosis, enhances diagnostic success. This study adds to our understanding of genotype-phenotype relationships in RASopathies and highlights new clinical features tied to SOS1 variants. Comprehensive genetic testing supports earlier and more personalized care for patients with RASopathies.
Publicações recentes
Novel Variants in PTPN11, NF1, RASA2, and MAP2K1: Expanding the Molecular Spectrum of RASopathies in a Turkish Cohort.
Molecular and Clinical Profiles of Patients with RASopathies: Targeted Next-Generation Sequencing Panel Results and Identification of 14 Novel Disease-Causing Variants.
Clinical and Genetic Characterization of Noonan Syndrome in a Romanian Cohort from Transylvania: Details on PTPN11 c.922A>G Variant and Phenotypic Spectrum.
Expanding the Genetic Landscape of RASopathies: Significance of Including NF1 in Targeted Panels.
[Analysis of clinical characteristics and NF1 gene variants in a child with Neurofibroma-Noonan syndrome].
📚 EuropePMC29 artigos no totalmostrando 20
Novel Variants in PTPN11, NF1, RASA2, and MAP2K1: Expanding the Molecular Spectrum of RASopathies in a Turkish Cohort.
Clinical geneticsMolecular and Clinical Profiles of Patients with RASopathies: Targeted Next-Generation Sequencing Panel Results and Identification of 14 Novel Disease-Causing Variants.
Molecular syndromologyClinical and Genetic Characterization of Noonan Syndrome in a Romanian Cohort from Transylvania: Details on PTPN11 c.922A>G Variant and Phenotypic Spectrum.
Diagnostics (Basel, Switzerland)Expanding the Genetic Landscape of RASopathies: Significance of Including NF1 in Targeted Panels.
Molecular syndromology[Analysis of clinical characteristics and NF1 gene variants in a child with Neurofibroma-Noonan syndrome].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsNeurofibromatosis-Noonan syndrome: a prospective monocentric study of 26 patients and literature review.
Orphanet journal of rare diseasesPortraying the full picture of Neurofibromatosis-Noonan syndrome: a systematic review of literature.
Journal of medical geneticsOrbital and Lumbosacral Plexiform Neurofibroma with PTPN11 Mutation: A Form of the RASopathy.
CureusA bicarotid trunk with associated right retroesophageal subclavian artery in a child with neurofibromatosis type 1 complicated by a left hemispheric stroke.
Surgical and radiologic anatomy : SRANeurofibromatosis-Noonan Syndrome With Primary Amenorrhoea: A Case Report.
CureusTurner Syndrome and Neurofibromatosis 1: Rare Co-Existence with Important Clinical Implications.
Journal of the ASEAN Federation of Endocrine SocietiesNeurofibromatosis-Noonan syndrome and growth deficiency in an Iranian girl due to a pathogenic variant in NF1 gene.
Human genomicsA Novel Heterozygous NF1 Variant in a Neurofibromatosis-Noonan Syndrome Patient with Growth Hormone Deficiency: A Case Report.
Journal of clinical research in pediatric endocrinologyExpanding the clinical phenotype of RASopathies in 38 Turkish patients, including the rare LZTR1, RAF1, RIT1 variants, and large deletion in NF1.
American journal of medical genetics. Part AChinese patient with neurofibromatosis-Noonan syndrome caused by novel heterozygous NF1 exons 1-58 deletion: a case report.
BMC pediatricsExpanding the Noonan spectrum/RASopathy NGS panel: Benefits of adding NF1 and SPRED1.
Molecular genetics & genomic medicineA Neurofibromatosis Noonan Syndrome Patient Presenting with Abnormal External Genitalia.
Journal of clinical research in pediatric endocrinologyNeurofibromatosis-Noonan Syndrome: A Possible Paradigm of the Combination of Genetic and Epigenetic Factors.
Advances in experimental medicine and biologyIs Neurofibromatosis Type 1-Noonan Syndrome a Phenotypic Result of Combined Genetic and Epigenetic Factors?
In vivo (Athens, Greece)Growth Hormone Deficiency in a Child with Neurofibromatosis-Noonan Syndrome.
Journal of clinical research in pediatric endocrinologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Associação brasileira dedicada a Doenças raras (geral).
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de neurofibromatose-Noonan
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Novel Variants in PTPN11, NF1, RASA2, and MAP2K1: Expanding the Molecular Spectrum of RASopathies in a Turkish Cohort.
- Neurofibromatosis-Noonan syndrome: a prospective monocentric study of 26 patients and literature review.
- Molecular and Clinical Profiles of Patients with RASopathies: Targeted Next-Generation Sequencing Panel Results and Identification of 14 Novel Disease-Causing Variants.
- Clinical and Genetic Characterization of Noonan Syndrome in a Romanian Cohort from Transylvania: Details on PTPN11 c.922A>G Variant and Phenotypic Spectrum.
- Expanding the Genetic Landscape of RASopathies: Significance of Including NF1 in Targeted Panels.
- [Analysis of clinical characteristics and NF1 gene variants in a child with Neurofibroma-Noonan syndrome].
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:638(Orphanet)
- OMIM OMIM:601321(OMIM)
- MONDO:0011035(MONDO)
- GARD:372(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q102293764(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
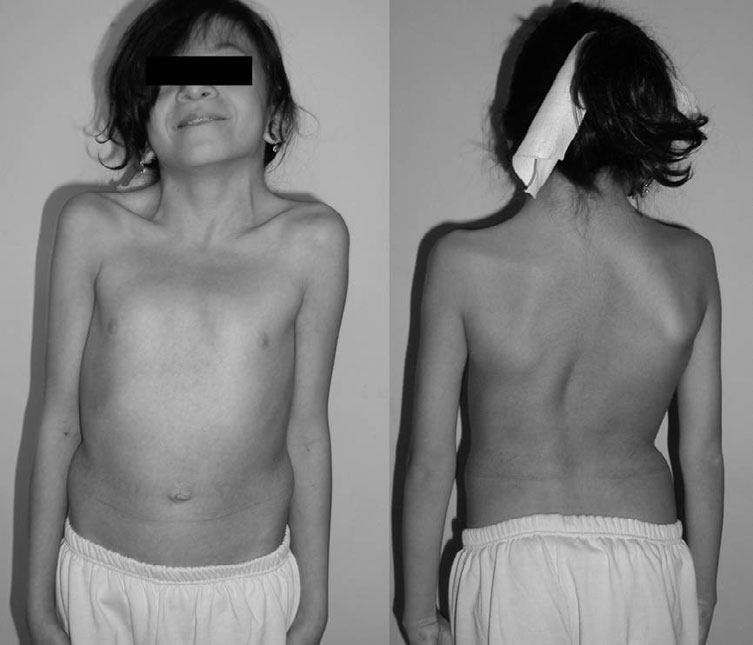
Síndrome de neurofibromatose-Noonan
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov