A doença de retenção de quilomícrons (DRC) é um tipo de hipocolesterolemia familiar caracterizada por desnutrição, retardo de crescimento, deficiência de crescimento, deficiência de vitamina E e complicações hepáticas, neurológicas e oftalmológicas.
Introdução
O que você precisa saber de cara
A doença de retenção de quilomícrons (DRC) é um tipo de hipocolesterolemia familiar caracterizada por desnutrição, retardo de crescimento, deficiência de crescimento, deficiência de vitamina E e complicações hepáticas, neurológicas e oftalmológicas.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 16 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 30 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisSmall GTPase that cycles between an active GTP-bound and an inactive GDP-bound state and mainly functions in vesicle-mediated endoplasmic reticulum (ER) to Golgi transport. The active GTP-bound form inserts into the endoplasmic reticulum membrane where it recruits the remainder of the coat protein complex II/COPII (PubMed:23433038, PubMed:32358066, PubMed:33186557, PubMed:36369712). The coat protein complex II assembling and polymerizing on endoplasmic reticulum membrane is responsible for both
Endoplasmic reticulum membraneGolgi apparatus, Golgi stack membraneCytoplasm, cytosolLysosome membrane
Chylomicron retention disease
An autosomal recessive disorder of severe fat malabsorption associated with failure to thrive in infancy. The condition is characterized by deficiency of fat-soluble vitamins, low blood cholesterol levels, and a selective absence of chylomicrons from blood. Affected individuals accumulate chylomicron-like particles in membrane-bound compartments of enterocytes, which contain large cytosolic lipid droplets.
Variantes genéticas (ClinVar)
55 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 17 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
7 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Doença de retenção dos quilomícrons
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
Pesquisa e ensaios clínicos
2 ensaios clínicos encontrados.
Publicações mais relevantes
Case Report: Neonatal-onset chylomicron retention disease presenting as isolated failure to thrive with compound heterozygous SAR1B variants: the value of early genetic testing and challenges of long-term management.
Chylomicron retention disease (CMRD) is a rare autosomal recessive disorder caused by pathogenic variants of SAR1B, in which defective intestinal chylomicron secretion leads to fat malabsorption, hypocholesterolemia, and failure to thrive in infancy. Its diagnosis is typically challenging because of its rarity, nonspecific early symptoms, and overlap with other metabolic and malabsorptive disorders. The present case is notable for its neonatal onset with isolated failure to gain weight and the absence of persistent diarrhea or steatorrhea, complicating early clinical suspicion. Initial metabolic screening revealed overlapping abnormalities with urea cycle disorders and fatty acid oxidation defects, underscoring diagnostic complexity. A definitive diagnosis was achieved through the identification of compound heterozygous likely pathogenic SAR1B variants, c.258G > A (p.Trp86Ter) and c.442C > T (p.Arg148Ter), thereby expanding the known phenotypic and genotypic spectrum of CMRD. The patient exhibited marked clinical and biochemical improvements following timely intervention with a low-fat, medium-chain triglyceride(MCT)-enriched diet and fat-soluble vitamin supplementation. However, subsequent follow-up revealed suboptimal adherence to the dietary regimen, leading to the emergence of typical steatorrhea, persistent growth failure, and neurodevelopmental delay, highlighting the critical and sustained role of strict nutritional management. This case highlights the importance of heightened clinical vigilance, timely genetic testing, and the necessity of ensuring long-term treatment adherence in infants presenting with unexplained growth failure and subtle hepatic abnormalities even when classic symptoms are absent. Taken together, this study provides valuable insights into the diagnostic challenges, therapeutic pitfalls, and management strategies of CMRD, emphasizing the need for multidisciplinary collaboration, enhanced awareness among clinicians, and further research to elucidate genotype-phenotype correlations and optimize patient care.
[Chylomicron retention disease caused by SAR1B gene variations in 2 cases and literatures review].
Objective: To summarize the genotype and clinical characteristics of chylomicron retention disease (CMRD) caused by secretion associated Ras related GTPase 1B (SAR1B) gene variations. Methods: Clinical data and genetic testing results of 2 children with CMRD treated at Children's Hospital of Fudan University and Jiangxi Provincial Children's Hospital from May 2022 to July 2023 were summarized. To provide an overview of the clinical and genetic characteristics of CMRD caused by SAR1B gene variations, all of the literature was searched and reviewed from China National Knowledge Infrastructure, Wanfang Data Knowledge Service Platform, China VIP database, China Biology Medicine disc and PubMed database (up to January 2024) with "chylomicron retention disease" "Anderson disease" or "Anderson syndrome" as the search terms. All relevant literatures were reviewed to summarize the clinical and genetic features of CMRD caused by SAR1B gene variations. Results: One 11-year-old boy and one 4-month-old girl with CMRD. Both patients had lipid malabsorption, failure to thrive, decreased cholesterol, elevated transaminase and creatine kinase, and Vitamin E deficiency, with homozygous variations (c.224A>G) and compound heterozygous variations (c.224A>G and c.554G>T) in SAR1B gene, respectively. Case 1 was followed up for over a month, and he still occasionally experienced lower limb muscle pain. Case 2 was followed up for more than a year, and her had caught up to normal levels. Both patients had no other significant discomfort. Literature search retrieved 0 Chinese literature and 22 English literatures. In addition to the 2 cases reported in this study, a total of 51 patients were identified as CMRD caused by SAR1B gene variations. Twenty-one types of SAR1B variants 10 missense, 4 nonsense, 3 frameshift, 1 in-frame deletion, 1 splice, 1 gross deletion, and 1 gross insertion-deletion were found among the 51 CMRD cases. Among all the patients, 49 cases had lipid malabsorption (43 cases had diarrhea or fatty diarrhea, 17 cases had vomiting, and 12 cases had abdominal distension), 45 cases had lipid soluble Vitamin deficiency (43 cases had Vitamin E deficiency, 10 cases had Vitamin A deficiency, 9 case had Vitamin D deficiency, and 5 cases had Vitamin K deficiency), 35 cases had failure to thrive, 32 cases had liver involvement (32 cases had elevated transaminases, 5 cases had fatty liver, and 3 cases had hepatomegaly), 29 cases had white small intestinal mucosa under endoscopy, and 17 cases had elevated creatine kinase, 14 cases had neuropathy, 5 cases had ocular lesions, 2 cases had acanthocytosis, 1 case had decreased cardiac ejection fraction, and 1 case was symptom-free. Conclusions: Early infancy failure to thrive and lipid malabsorption are common issues for CMRD patients. The laboratory tests are characterized by hypocholesterolemia with or without fat-soluble Vitamin deficiency, elevated liver enzymes and (or) creatine kinase. Currently, missense variations are frequent among the primarily homozygous SAR1B genotypes that have been described. 目的: 总结SAR1B基因变异致乳糜微粒滞留病(CMRD)的基因型与临床特点。 方法: 病例系列研究,总结2022年5月至2023年7月复旦大学附属儿科医院与江西省儿童医院诊治的2例CMRD患儿的临床资料与基因检测结果。分别以“乳糜微粒滞留病”“乳糜微粒潴留病”“乳糜微粒保留病”“安德森病”“安德森综合征”“chylomicron retention disease”“Anderson syndrome”“Anderson disease”为检索词在中国知网、万方、维普、中国生物医学文献、PubMed数据库自建库至2024年1月进行检索。总结SAR1B基因变异致CMRD的主要临床表现及遗传学特点。 结果: 2例患儿分别为1例11岁男童与1例4月龄女童。2例患儿均有脂质吸收不良、生长不良、胆固醇下降、转氨酶与肌酸激酶升高、维生素E缺乏,基因检查发现分别为SAR1B基因纯合变异(c.224A>G)和复合杂合变异(c.224A>G和c.554G>T)。例1随访1个月余,仍偶有下肢肌痛;例2随访1年余,生长已追赶至正常水平,2例患儿均无其他明显不适。文献复习符合检索条件中文文献0篇,英文文献22篇,包括本研究2例共有51例患者。51例CMRD患者中存在21种SAR1B基因变异,包含10种错义变异,4种无义变异,3种移码变异,框内缺失、剪接位点变异、大片段缺失与大片段缺失插入各1种。51例CMRD患者中,脂质吸收不良49例(腹泻或脂肪泻43例、呕吐17例、腹胀12例)、脂溶性维生素缺乏45例(维生素E缺乏43例、维生素A缺乏10例、维生素D缺乏9例、维生素K缺乏5例),生长不良35例,肝脏受累32例(转氨酶升高32例、脂肪肝5例、肝肿大3例),内镜下可见白色小肠黏膜29例,肌酸激酶升高17例,神经病变14例,眼部病变5例,棘形红细胞增多2例,心脏射血分数降低1例,无症状1例。 结论: CMRD常表现为婴儿早期脂质吸收不良和生长不良。实验室检测特点为低胆固醇血症,伴或不伴脂溶性维生素缺乏、肝酶和(或)肌酸激酶升高。SAR1B基因型以纯合型为主,错义变异多见。.
Chylomicron retention disease: a rare aetiology of failure to thrive.
The aetiology of failure to thrive (FTT) in children is broad, of which some conditions are extremely rare. It is important to consider these rarer conditions, especially in the setting of other concerning signs/symptoms or when there is no improvement with conventional treatment. In this case report we highlight such a rare condition-chylomicron retention disease (CRD) as an aetiology of FTT. CRD often presents with non-specific symptoms, resulting in delayed diagnosis which is established by genetic workup and histology from small intestinal biopsies. Despite being rare, CRD needs to be considered as one of the differential diagnoses after ruling out the more common causes of FTT.
Functional overlap between the mammalian Sar1a and Sar1b paralogs in vivo.
Proteins carrying a signal peptide and/or a transmembrane domain enter the intracellular secretory pathway at the endoplasmic reticulum (ER) and are transported to the Golgi apparatus via COPII vesicles or tubules. SAR1 initiates COPII coat assembly by recruiting other coat proteins to the ER membrane. Mammalian genomes encode two SAR1 paralogs, SAR1A and SAR1B. While these paralogs exhibit ~90% amino acid sequence identity, it is unknown whether they perform distinct or overlapping functions in vivo. We now report that genetic inactivation of Sar1a in mice results in lethality during midembryogenesis. We also confirm previous reports that complete deficiency of murine Sar1b results in perinatal lethality. In contrast, we demonstrate that deletion of Sar1b restricted to hepatocytes is compatible with survival, though resulting in hypocholesterolemia that can be rescued by adenovirus-mediated overexpression of either SAR1A or SAR1B. To further examine the in vivo function of these two paralogs, we genetically engineered mice with the Sar1a coding sequence replacing that of Sar1b at the endogenous Sar1b locus. Mice homozygous for this allele survive to adulthood and are phenotypically normal, demonstrating complete or near-complete overlap in function between the two SAR1 protein paralogs in mice. These data also suggest upregulation of SAR1A gene expression as a potential approach for the treatment of SAR1B deficiency (chylomicron retention disease) in humans.
Unraveling Chylomicron Retention Disease Enhances Insight into SAR1B GTPase Functions and Mechanisms of Actions, While Shedding Light of Intracellular Chylomicron Trafficking.
Over the past three decades, significant efforts have been focused on unraveling congenital intestinal disorders that disrupt the absorption of dietary lipids and fat-soluble vitamins. The primary goal has been to gain deeper insights into intra-enterocyte sites, molecular steps, and crucial proteins/regulatory pathways involved, while simultaneously identifying novel therapeutic targets and diagnostic tools. This research not only delves into specific and rare malabsorptive conditions, such as chylomicron retention disease (CRD), but also contributes to our understanding of normal physiology through the utilization of cutting-edge cellular and animal models alongside advanced research methodologies. This review elucidates how modern techniques have facilitated the decoding of CRD gene defects, the identification of dysfunctional cellular processes, disease regulatory mechanisms, and the essential role of coat protein complex II-coated vesicles and cargo receptors in chylomicron trafficking and endoplasmic reticulum (ER) exit sites. Moreover, experimental approaches have shed light on the multifaceted functions of SAR1B GTPase, wherein loss-of-function mutations not only predispose individuals to CRD but also exacerbate oxidative stress, inflammation, and ER stress, potentially contributing to clinical complications associated with CRD. In addition to dissecting the primary disease pathology, genetically modified animal models have emerged as invaluable assets in exploring various ancillary aspects, including responses to environmental challenges such as dietary alterations, gender-specific disparities in disease onset and progression, and embryonic lethality or developmental abnormalities. In summary, this comprehensive review provides an in-depth and contemporary analysis of CRD, offering a meticulous examination of the CRD current landscape by synthesizing the latest research findings and advancements in the field.
Publicações recentes
Case Report: Neonatal-onset chylomicron retention disease presenting as isolated failure to thrive with compound heterozygous SAR1B variants: the value of early genetic testing and challenges of long-term management.
Chylomicron retention disease: A condition to keep in mind.
Unraveling Chylomicron Retention Disease Enhances Insight into SAR1B GTPase Functions and Mechanisms of Actions, While Shedding Light of Intracellular Chylomicron Trafficking.
[Chylomicron retention disease caused by SAR1B gene variations in 2 cases and literatures review].
Chylomicron retention disease: a rare aetiology of failure to thrive.
📚 EuropePMC45 artigos no totalmostrando 36
Case Report: Neonatal-onset chylomicron retention disease presenting as isolated failure to thrive with compound heterozygous SAR1B variants: the value of early genetic testing and challenges of long-term management.
Frontiers in pediatricsChylomicron retention disease: A condition to keep in mind.
Anales de pediatriaUnraveling Chylomicron Retention Disease Enhances Insight into SAR1B GTPase Functions and Mechanisms of Actions, While Shedding Light of Intracellular Chylomicron Trafficking.
Biomedicines[Chylomicron retention disease caused by SAR1B gene variations in 2 cases and literatures review].
Zhonghua er ke za zhi = Chinese journal of pediatricsChylomicron retention disease: a rare aetiology of failure to thrive.
BMJ case reportsFunctional overlap between the mammalian Sar1a and Sar1b paralogs in vivo.
Proceedings of the National Academy of Sciences of the United States of AmericaCarotenoids in familial hypobetalipoproteinemia disorders: Malabsorption in Caco2 cell models and severe deficiency in patients.
Journal of clinical lipidologyHigh-fat diet reveals the impact of Sar1b defects on lipid and lipoprotein profile and cholesterol metabolism.
Journal of lipid researchLow cholesterol states: clinical implications and management.
Expert review of endocrinology & metabolismValidation of Knock-Out Caco-2 TC7 Cells as Models of Enterocytes of Patients with Familial Genetic Hypobetalipoproteinemias.
NutrientsGuidance for the diagnosis and treatment of hypolipidemia disorders.
Journal of clinical lipidologyPhenotypic Findings in Patients With KCNJ2-Related Anderson-Tawil Syndrome.
NeurologyCongenital disorders of intestinal digestion and absorption (sugars, proteins, lipids, ions).
Best practice & research. Clinical gastroenterologyChylomicron Retention Disease: Failure to Thrive and Abdominal Distention in an Infant.
JPGN reportsChylomicron retention disease caused by a new pathogenic variant in sar1b protein: a rare case report from Syria.
BMC pediatricsSar1b mutant mice recapitulate gastrointestinal abnormalities associated with chylomicron retention disease.
Journal of lipid researchLipids Responsible for Intestinal or Hepatic Disorder: When to Suspect a Familial Intestinal Hypocholesterolemia?
Journal of pediatric gastroenterology and nutritionNew Classification and Management of Abetalipoproteinemia and Related Disorders.
GastroenterologyInhibition of Sar1b, the Gene Implicated in Chylomicron Retention Disease, Impairs Migration and Morphogenesis of Developing Cortical Neurons.
NeuroscienceEffects of agalsidase-β administration on vascular function and blood pressure in familial Anderson-Fabry disease.
European journal of human genetics : EJHGChylomicron Retention Disease in A Male Infant: A Rare Case from Pakistan.
CureusNovel mutations of SAR1B gene in four children with chylomicron retention disease.
Journal of clinical lipidologyMolecular analysis of APOB, SAR1B, ANGPTL3, and MTTP in patients with primary hypocholesterolemia in a clinical laboratory setting: Evidence supporting polygenicity in mutation-negative patients.
AtherosclerosisChylomicron retention disease: genetics, biochemistry, and clinical spectrum.
Current opinion in lipidologyEfficacy of two vitamin E formulations in patients with abetalipoproteinemia and chylomicron retention disease.
Journal of lipid researchChylomicron Retention Disease: a Description of a New Mutation in a Very Rare Disease.
Pediatric gastroenterology, hepatology & nutritionComplex genetic architecture in severe hypobetalipoproteinemia.
Lipids in health and diseaseUnderstanding Chylomicron Retention Disease Through Sar1b Gtpase Gene Disruption: Insight From Cell Culture.
Arteriosclerosis, thrombosis, and vascular biologyUnderstanding Plastid Vesicle Transport - Could it Provide Benefit for Human Medicine?
Mini reviews in medicinal chemistryEstablishment of reference values of α-tocopherol in plasma, red blood cells and adipose tissue in healthy children to improve the management of chylomicron retention disease, a rare genetic hypocholesterolemia.
Orphanet journal of rare diseasesChylomicron retention disease: A rare cause of chronic diarrhea.
Archives de pediatrie : organe officiel de la Societe francaise de pediatrieChylomicrons: Advances in biology, pathology, laboratory testing, and therapeutics.
Clinica chimica acta; international journal of clinical chemistryHomozygous familial hypobetalipoproteinemia: A Turkish case carrying a missense mutation in apolipoprotein B.
Clinica chimica acta; international journal of clinical chemistryUpdate on the molecular biology of dyslipidemias.
Clinica chimica acta; international journal of clinical chemistryStudying lipoprotein trafficking in zebrafish, the case of chylomicron retention disease.
Journal of molecular medicine (Berlin, Germany)Animal model of Sar1b deficiency presents lipid absorption deficits similar to Anderson disease.
Journal of molecular medicine (Berlin, Germany)Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Doença de retenção dos quilomícrons.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Doença de retenção dos quilomícrons
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Case Report: Neonatal-onset chylomicron retention disease presenting as isolated failure to thrive with compound heterozygous SAR1B variants: the value of early genetic testing and challenges of long-term management.
- [Chylomicron retention disease caused by SAR1B gene variations in 2 cases and literatures review].
- Chylomicron retention disease: a rare aetiology of failure to thrive.
- Functional overlap between the mammalian Sar1a and Sar1b paralogs in vivo.Proceedings of the National Academy of Sciences of the United States of America· 2024· PMID 38687799mais citado
- Unraveling Chylomicron Retention Disease Enhances Insight into SAR1B GTPase Functions and Mechanisms of Actions, While Shedding Light of Intracellular Chylomicron Trafficking.
- Chylomicron retention disease: A condition to keep in mind.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:71(Orphanet)
- OMIM OMIM:246700(OMIM)
- MONDO:0009528(MONDO)
- GARD:9683(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q959211(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
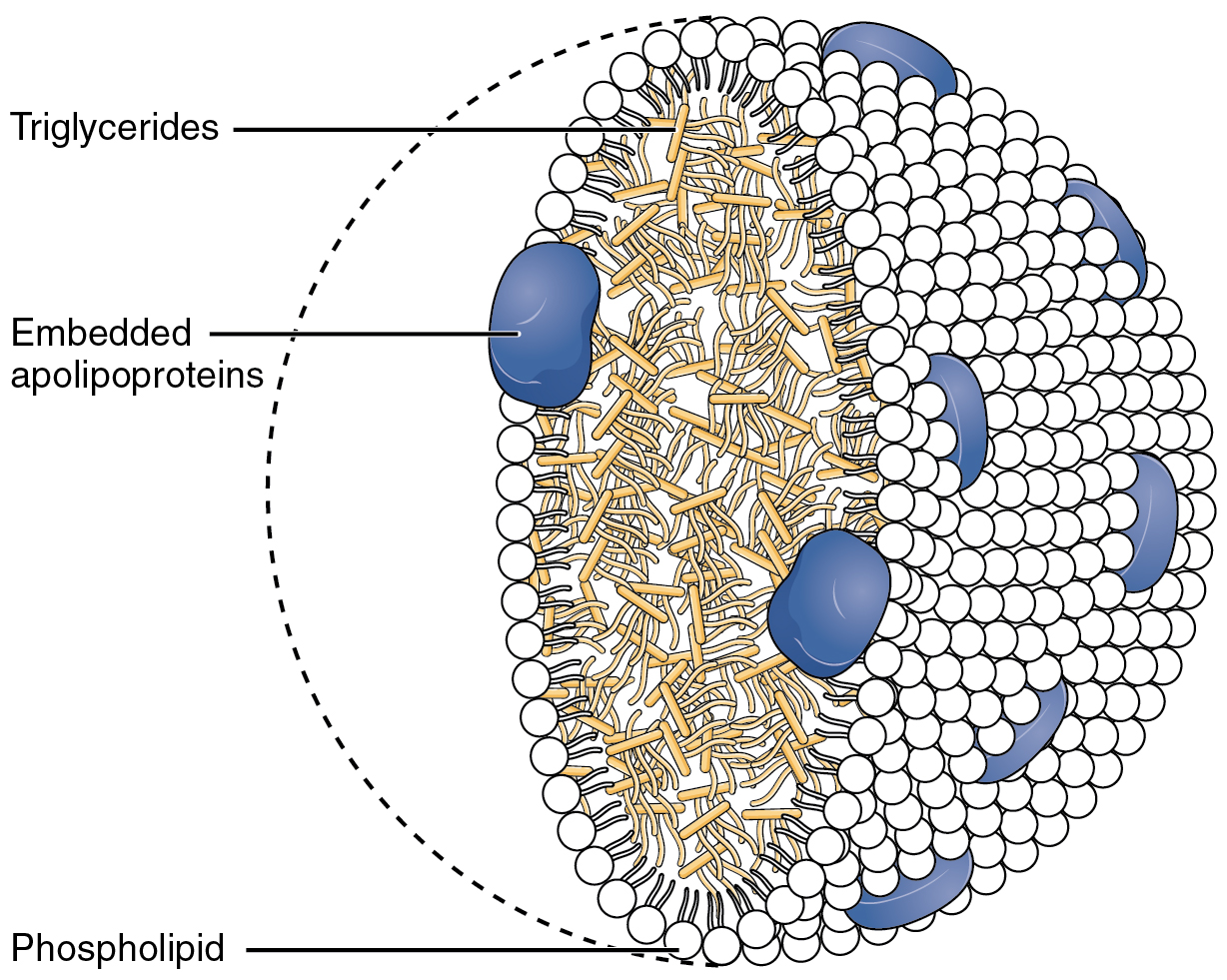
Doença de retenção dos quilomícrons
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata