Cegueira noturna estacionária congênita é uma condição genética rara que causa dificuldade de enxergar em ambientes com pouca luz desde o nascimento. Pode apresentar visão reduzida, estrabismo, alterações na pigmentação da retina e defeitos na visão de cores, com herança autossômica ou ligada ao X.
Introdução
O que você precisa saber de cara
Visão geral
A cegueira noturna estacionária congênita é uma doença hereditária da retina que afeta a visão desde o nascimento. A principal característica é a dificuldade de enxergar em ambientes com pouca luz (nictalopia), que não piora com o tempo — por isso é chamada de 'estacionária'. A condição pode se apresentar com ou sem alterações visíveis no fundo do olho ao exame oftalmológico.[1][3]
A prevalência exata da doença é desconhecida. Ela pode ser herdada de três formas diferentes: autossômica dominante, autossômica recessiva ou ligada ao cromossomo X. Os sintomas estão presentes desde o período neonatal.[1][3]
Sinais e sintomas
O sintoma mais marcante é a cegueira noturna (nictalopia) — dificuldade para enxergar em ambientes escuros ou com iluminação fraca. Em alguns casos, a pessoa também pode ter dificuldade para enxergar durante o dia (hemeralopia) e sensibilidade à luz (fotofobia).[1][3]
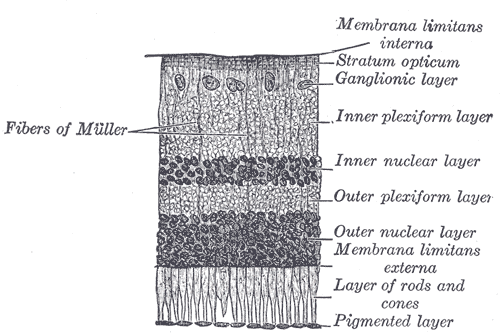
Outros sinais frequentes incluem: acuidade visual reduzida (visão embaçada), miopia (especialmente alta miopia), hipermetropia, estrabismo (desalinhamento dos olhos), nistagmo (movimentos involuntários dos olhos) e alterações na visão de cores. O exame de eletrorretinograma costuma mostrar um padrão característico chamado 'eletronegativo', com redução da amplitude da onda A na adaptação ao escuro.[1][3]
Em algumas pessoas, o fundo do olho pode apresentar alterações como adelgaçamento retiniano, anormalidades na pigmentação ou o chamado 'Fenômeno de Mizuo' (mudança na cor da retina após exposição à luz). Em outras, o fundo do olho pode ser completamente normal.[1][3]
Causas genéticas
A cegueira noturna estacionária congênita é causada por alterações (mutações) em genes essenciais para o funcionamento dos bastonetes, as células da retina responsáveis pela visão em baixa luminosidade. Atualmente, mais de 12 genes estão associados à doença, incluindo: GPR179, PDE6B, CABP4, GUCY2D, TRPM1, GNAT1, NYX, GRM6, GRK1, SAG, RHO e LRIT3.[1][4]
Cada gene tem um papel específico na cascata de sinalização visual. Por exemplo, o gene RHO codifica a rodopsina, proteína fundamental para a detecção da luz. Mutações em diferentes genes podem levar a formas ligeiramente distintas da doença, mas todas compartilham a característica de serem congênitas e estacionárias.[1][4]
Diagnóstico
O diagnóstico é baseado na história clínica (sintomas desde o nascimento), exame oftalmológico completo e exames complementares. O eletrorretinograma é fundamental para confirmar o padrão típico da doença. Exames de imagem da retina e testes de visão de cores também auxiliam na avaliação.[1][3]
O teste genético molecular pode identificar a mutação causadora em um dos genes associados. Atualmente, há mais de 500 variantes genéticas descritas em bases de dados como o ClinVar, e a taxa de sucesso do teste genético é estimada em 80%. O diagnóstico genético é importante para confirmar o tipo de herança e orientar o aconselhamento familiar.[1][4][5]
Tratamento e manejo
Atualmente, não existe cura para a cegueira noturna estacionária congênita. O tratamento é focado no manejo dos sintomas e na melhora da qualidade de vida. O uso de óculos ou lentes de contato pode corrigir a miopia ou hipermetropia associadas. Em casos de estrabismo ou nistagmo significativo, a cirurgia ou terapia visual podem ser consideradas.[1][5]
Recomenda-se o acompanhamento regular com oftalmologista especializado em retina e genética ocular. Medidas de baixa visão, como o uso de lupas e dispositivos de ampliação, podem ajudar nas atividades diárias. A proteção contra luz excessiva (óculos escuros) pode aliviar a fotofobia.[1][5]
Tratamentos citados na literatura
A literatura científica (fonte: PubTator3) menciona algumas substâncias em estudos sobre a doença, mas é importante destacar que estas são associações mineradas de publicações científicas e NÃO representam recomendações de tratamento. As substâncias com maior número de publicações associadas são: Cálcio (26 publicações), Melatonina (14), Lipídios (8), Colesterol (6), Dopamina (6), Ácido Glutâmico (6), Espécies Reativas de Oxigênio (6), Fluordesoxiglicose F18 (5), Óxido Nítrico (5) e Licopeno (5).[5]
Prognóstico e qualidade de vida
O prognóstico visual é geralmente estável, já que a doença não é progressiva. A maioria das pessoas mantém a visão que tem desde a infância, sem piora significativa ao longo da vida. A principal limitação é a dificuldade em ambientes com pouca luz, o que pode exigir adaptações no dia a dia, como evitar dirigir à noite.[1][3]
Com o suporte adequado (óculos, auxílios ópticos e acompanhamento médico), a maioria das pessoas consegue levar uma vida independente e produtiva. O aconselhamento genético é recomendado para famílias afetadas.[1][5]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Cegueira noturna estacionária congênita é uma condição genética rara que causa dificuldade de enxergar em ambientes com pouca luz desde o nascimento. Pode apresentar visão reduzida, estrabismo, alterações na pigmentação da retina e defeitos na visão de cores, com herança autossômica ou ligada ao X.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A cegueira noturna estacionária congênita é uma doença hereditária da retina que afeta a visão desde o nascimento. A principal característica é a dificuldade de enxergar em ambientes com pouca luz (nictalopia), que não piora com o tempo — por isso é chamada de 'estacionária'. A condição pode se apresentar com ou sem alterações visíveis no fundo do olho ao exame oftalmológico.[1][3]
A prevalência exata da doença é desconhecida. Ela pode ser herdada de três formas diferentes: autossômica dominante, autossômica recessiva ou ligada ao cromossomo X. Os sintomas estão presentes desde o período neonatal.[1][3]
Sinais e sintomas
O sintoma mais marcante é a cegueira noturna (nictalopia) — dificuldade para enxergar em ambientes escuros ou com iluminação fraca. Em alguns casos, a pessoa também pode ter dificuldade para enxergar durante o dia (hemeralopia) e sensibilidade à luz (fotofobia).[1][3]
Outros sinais frequentes incluem: acuidade visual reduzida (visão embaçada), miopia (especialmente alta miopia), hipermetropia, estrabismo (desalinhamento dos olhos), nistagmo (movimentos involuntários dos olhos) e alterações na visão de cores. O exame de eletrorretinograma costuma mostrar um padrão característico chamado 'eletronegativo', com redução da amplitude da onda A na adaptação ao escuro.[1][3]
Em algumas pessoas, o fundo do olho pode apresentar alterações como adelgaçamento retiniano, anormalidades na pigmentação ou o chamado 'Fenômeno de Mizuo' (mudança na cor da retina após exposição à luz). Em outras, o fundo do olho pode ser completamente normal.[1][3]
Causas genéticas
A cegueira noturna estacionária congênita é causada por alterações (mutações) em genes essenciais para o funcionamento dos bastonetes, as células da retina responsáveis pela visão em baixa luminosidade. Atualmente, mais de 12 genes estão associados à doença, incluindo: GPR179, PDE6B, CABP4, GUCY2D, TRPM1, GNAT1, NYX, GRM6, GRK1, SAG, RHO e LRIT3.[1][4]
Cada gene tem um papel específico na cascata de sinalização visual. Por exemplo, o gene RHO codifica a rodopsina, proteína fundamental para a detecção da luz. Mutações em diferentes genes podem levar a formas ligeiramente distintas da doença, mas todas compartilham a característica de serem congênitas e estacionárias.[1][4]
Diagnóstico
O diagnóstico é baseado na história clínica (sintomas desde o nascimento), exame oftalmológico completo e exames complementares. O eletrorretinograma é fundamental para confirmar o padrão típico da doença. Exames de imagem da retina e testes de visão de cores também auxiliam na avaliação.[1][3]
O teste genético molecular pode identificar a mutação causadora em um dos genes associados. Atualmente, há mais de 500 variantes genéticas descritas em bases de dados como o ClinVar, e a taxa de sucesso do teste genético é estimada em 80%. O diagnóstico genético é importante para confirmar o tipo de herança e orientar o aconselhamento familiar.[1][4][5]
Tratamento e manejo
Atualmente, não existe cura para a cegueira noturna estacionária congênita. O tratamento é focado no manejo dos sintomas e na melhora da qualidade de vida. O uso de óculos ou lentes de contato pode corrigir a miopia ou hipermetropia associadas. Em casos de estrabismo ou nistagmo significativo, a cirurgia ou terapia visual podem ser consideradas.[1][5]
Recomenda-se o acompanhamento regular com oftalmologista especializado em retina e genética ocular. Medidas de baixa visão, como o uso de lupas e dispositivos de ampliação, podem ajudar nas atividades diárias. A proteção contra luz excessiva (óculos escuros) pode aliviar a fotofobia.[1][5]
Tratamentos citados na literatura
A literatura científica (fonte: PubTator3) menciona algumas substâncias em estudos sobre a doença, mas é importante destacar que estas são associações mineradas de publicações científicas e NÃO representam recomendações de tratamento. As substâncias com maior número de publicações associadas são: Cálcio (26 publicações), Melatonina (14), Lipídios (8), Colesterol (6), Dopamina (6), Ácido Glutâmico (6), Espécies Reativas de Oxigênio (6), Fluordesoxiglicose F18 (5), Óxido Nítrico (5) e Licopeno (5).[5]
Prognóstico e qualidade de vida
O prognóstico visual é geralmente estável, já que a doença não é progressiva. A maioria das pessoas mantém a visão que tem desde a infância, sem piora significativa ao longo da vida. A principal limitação é a dificuldade em ambientes com pouca luz, o que pode exigir adaptações no dia a dia, como evitar dirigir à noite.[1][3]
Com o suporte adequado (óculos, auxílios ópticos e acompanhamento médico), a maioria das pessoas consegue levar uma vida independente e produtiva. O aconselhamento genético é recomendado para famílias afetadas.[1][5]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 12 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 45 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
15 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive, X-linked recessive.
Orphan receptor involved in vision (PubMed:22325362, PubMed:24084093). Required for signal transduction through retinal depolarizing bipolar cells (PubMed:22325362). Acts as an atypical G-protein coupled receptor that recruits and regulates the R7 group RGS-GNB5 complexes instead of activating G proteins: promotes the GTPase activator activity of R7 RGS proteins, increasing the GTPase activity of G protein alpha subunits, thereby driving them into their inactive GDP-bound form (By similarity). A
Cell membranePostsynaptic cell membraneCell projection, dendrite
Night blindness, congenital stationary, 1E
An autosomal recessive, non-progressive retinal disorder characterized by impaired night vision, absence of the electroretinogram (ERG) b-wave, and variable degrees of involvement of other visual functions. Affected individuals have an ERG waveform that lacks the b-wave because of failure to transmit the photoreceptor signal through the retinal depolarizing bipolar cells.
Rod-specific cGMP phosphodiesterase that catalyzes the hydrolysis of 3',5'-cyclic GMP (PubMed:20940301). Necessary for the formation of a functional phosphodiesterase holoenzyme (By similarity). Involved in retinal circadian rhythm photoentrainment via modulation of UVA and orange light-induced phase-shift of the retina clock (By similarity). May participate in processes of transmission and amplification of the visual signal (PubMed:8394174)
MembraneCell projection, cilium, photoreceptor outer segment
Retinitis pigmentosa 40
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Involved in normal synaptic function through regulation of Ca(2+) influx and neurotransmitter release in photoreceptor synaptic terminals and in auditory transmission. Modulator of CACNA1D and CACNA1F, suppressing the calcium-dependent inactivation and shifting the activation range to more hyperpolarized voltages (By similarity)
CytoplasmPresynapse
Cone-rod synaptic disorder, congenital non-progressive
A non-progressive retinal disorder characterized by stable low vision, nystagmus, photophobia, a normal or near-normal fundus appearance, and no night blindness.
Catalyzes the synthesis of cyclic GMP (cGMP) in rods and cones of photoreceptors. Plays an essential role in phototransduction, by mediating cGMP replenishment (PubMed:15123990, PubMed:21928830, PubMed:26100624, PubMed:30319355, PubMed:9600905). May also participate in the trafficking of membrane-associated proteins to the photoreceptor outer segment membrane (By similarity)
Photoreceptor outer segment membraneEndoplasmic reticulum membrane
Leber congenital amaurosis 1
A severe dystrophy of the retina, typically becoming evident in the first years of life. Visual function is usually poor and often accompanied by nystagmus, sluggish or near-absent pupillary responses, photophobia, high hyperopia and keratoconus.
Constitutively open nonselective divalent cation-conducting channels which mediate the influx of Ca(2+), Mg(2+), Mn(2+), Ba(2+), and Ni(2+) into the cytoplasm, leading to membrane depolarization (PubMed:11535825, PubMed:19436059, PubMed:21278253). Impermeable to zinc ions (PubMed:21278253). In addition, forms heteromultimeric ion channels with TRPM3 which are permeable for calcium and zinc ions (PubMed:21278253). Plays an essential role for the depolarizing photoresponse of retinal ON bipolar ce
Cell membraneEndoplasmic reticulum membraneCell projection, axon
Night blindness, congenital stationary, 1C
A non-progressive retinal disorder characterized by impaired night vision, often associated with nystagmus and myopia.
Functions as a signal transducer for the rod photoreceptor RHO. Required for normal RHO-mediated light perception by the retina (PubMed:22190596). Guanine nucleotide-binding proteins (G proteins) function as transducers downstream of G protein-coupled receptors (GPCRs), such as the photoreceptor RHO. The alpha chain contains the guanine nucleotide binding site and alternates between an active, GTP-bound state and an inactive, GDP-bound state. Activated RHO promotes GDP release and GTP binding. S
Cell projection, cilium, photoreceptor outer segmentMembranePhotoreceptor inner segment
Night blindness, congenital stationary, autosomal dominant 3
A non-progressive retinal disorder characterized by impaired night vision, often associated with nystagmus and myopia.
Required for normal vision. Is a critical factor for light-induced depolarization of retinal ON-bipolar cells, likely acting as a scaffold for TRPM1 and GRM6. Required for TRPM1 trafficking to dendritic tips of ON-bipolar cells
Secreted, extracellular space, extracellular matrixCell projection, dendritePostsynapse
Night blindness, congenital stationary, 1A
A non-progressive retinal disorder characterized by impaired night vision. Congenital stationary night blindness type 1A is characterized by impaired scotopic vision, myopia, hyperopia, nystagmus and reduced visual acuity.
G-protein coupled receptor for glutamate. Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors, such as adenylate cyclase. Signaling inhibits adenylate cyclase activity (By similarity). Signaling stimulates TRPM1 channel activity and Ca(2+) uptake. Required for normal vision
Cell membraneEndoplasmic reticulum membraneGolgi apparatus membraneCell projection, dendrite
Night blindness, congenital stationary, 1B
A non-progressive retinal disorder characterized by impaired night vision. Congenital stationary night blindness type 1B is an autosomal recessive form associated with a negative electroretinogram waveform. Patients are night blind from an early age, and when maximally dark-adapted, they could perceive lights only with an intensity equal to or slightly dimmer than that normally detected by the cone system. ERGs in response to single brief flashes of light have clearly detectable a-waves, which are derived from photoreceptors, and greatly reduced b-waves, which are derived from the second-order inner retinal neurons. ERGs in response to sawtooth flickering light indicate a markedly reduced on response and a nearly normal OFF response. There is no subjective delay in the perception of suddenly appearing white vs black objects on a gray background.
Retina-specific kinase involved in the signal turnoff via phosphorylation of rhodopsin (RHO), the G protein- coupled receptor that initiates the phototransduction cascade (PubMed:15946941). This rapid desensitization is essential for scotopic vision and permits rapid adaptation to changes in illumination (By similarity). May play a role in the maintenance of the outer nuclear layer in the retina (By similarity)
MembraneCell projection, cilium, photoreceptor outer segment
Night blindness, congenital stationary, Oguchi type 2
A non-progressive retinal disorder characterized by impaired night vision, often associated with nystagmus and myopia. Congenital stationary night blindness Oguchi type is associated with fundus discoloration and abnormally slow dark adaptation.
Binds to photoactivated, phosphorylated RHO and terminates RHO signaling via G-proteins by competing with G-proteins for the same binding site on RHO (By similarity). May play a role in preventing light-dependent degeneration of retinal photoreceptor cells (PubMed:9565049)
Cell projection, cilium, photoreceptor outer segmentMembrane
Night blindness, congenital stationary, Oguchi type 1
A non-progressive retinal disorder characterized by impaired night vision, often associated with nystagmus and myopia. Congenital stationary night blindness Oguchi type is an autosomal recessive form associated with fundus discoloration and abnormally slow dark adaptation.
Photoreceptor required for image-forming vision at low light intensity (PubMed:7846071, PubMed:8107847). Required for photoreceptor cell viability after birth (PubMed:12566452, PubMed:2215617). Light-induced isomerization of the chromophore 11-cis-retinal to all-trans-retinal triggers a conformational change that activates signaling via G-proteins (PubMed:26200343, PubMed:28524165, PubMed:28753425, PubMed:8107847). Subsequent receptor phosphorylation mediates displacement of the bound G-protein
MembraneCell projection, cilium, photoreceptor outer segment
Retinitis pigmentosa 4
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Plays a role in the synapse formation and synaptic transmission between cone photoreceptor cells and retinal bipolar cells (By similarity). Required for normal transmission of a light-evoked stimulus from the cone photoreceptor cells to the ON-bipolar cells and ON-ganglion cells in the inner retina (PubMed:28334377). Required in retinal ON-bipolar cells for normal localization of the cation channel TRPM1 at dendrite tips (By similarity). Seems to play a specific role in synaptic contacts made by
Cell projection, dendritePerikaryonEndoplasmic reticulum membrane
Night blindness, congenital stationary, 1F
An autosomal recessive form of congenital stationary night blindness, a non-progressive retinal disorder characterized by impaired night vision, often associated with nystagmus and myopia.
Guanine nucleotide-binding proteins (G proteins) are involved as a modulator or transducer in various transmembrane signaling systems. The beta and gamma chains are required for the GTPase activity, for replacement of GDP by GTP, and for G protein-effector interaction
Night blindness, congenital stationary, 1H
A form of congenital stationary night blindness, a non-progressive retinal disorder characterized by impaired night vision or in dim light, with good vision only on bright days. CSNB1H patients present with childhood-onset night blindness and middle age-onset photophobia, but have near-normal vision and do not exhibit nystagmus or high myopia. CSNB1H inheritance is autosomal recessive.
Voltage-sensitive calcium channels (VSCC) mediate the entry of calcium ions into excitable cells and are also involved in a variety of calcium-dependent processes, including muscle contraction, hormone or neurotransmitter release, gene expression, cell motility, cell division and cell death. The isoform alpha-1F gives rise to L-type calcium currents. Long-lasting (L-type) calcium channels belong to the 'high-voltage activated' (HVA) group. They are blocked by dihydropyridines (DHP), phenylalkyla
Membrane
Night blindness, congenital stationary, 2A
A non-progressive retinal disorder characterized by impaired night vision, often associated with nystagmus and myopia.
Calcium, potassium:sodium antiporter that transports 1 Ca(2+) and 1 K(+) in exchange for 4 Na(+) (PubMed:26631410). Critical component of the visual transduction cascade, controlling the calcium concentration of outer segments during light and darkness (PubMed:20850105). Light causes a rapid lowering of cytosolic free calcium in the outer segment of both retinal rod and cone photoreceptors and the light-induced lowering of calcium is caused by extrusion via this protein which plays a key role in
Cell membrane
Night blindness, congenital stationary, 1D
An autosomal recessive form of congenital stationary night blindness, a non-progressive retinal disorder characterized by impaired night vision. CSNB1D is characterized by a Riggs type of electroretinogram (proportionally reduced a- and b-waves). Patients have visual acuity within the normal range and no symptoms of myopia and/or nystagmus.
Variantes genéticas (ClinVar)
513 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 1.072 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
40 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Cegueira noturna estacionária congênita
Centros de Referência SUS
24 centros habilitados pelo SUS para Cegueira noturna estacionária congênita
Centros para Cegueira noturna estacionária congênita
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
3 ensaios clínicos encontrados, 1 ativos.
Publicações mais relevantes
Cryo-EM structure of TRPM1 reveals a non-canonical architecture with an inverted transmembrane domain.
Transient receptor potential melastatin 1 (TRPM1) is a membrane protein essential for vision in dim light, and mutations in TRPM1 cause complete congenital stationary night blindness. Although TRPM1 shares sequence similarity to other TRPM ion channels such as TRPM3, whether it independently functions as an ion channel remains controversial. This controversy is largely caused by TRPM1's challenging biochemical behaviors that prevent detailed molecular characterization. In this work, we isolate TRPM1 and determine its structures using cryogenic electron microscopy (cryo-EM). The structures reveal a canonical tetrameric fold in the intracellular domain, consistent with other TRPM family members that are ion channels. Surprisingly, in the transmembrane domain, despite the presence of the conserved voltage sensor-like domain (VSLD) and pore domain (PD) in a domain-swapped fashion, the VSLD and PD are arranged with an opposite handedness compared to other related channels. This inverted transmembrane domain allows the formation of a large pore-like structure that supports the role of TRPM1 as an ion channel. This non-canonical architecture of TRPM1 may also confer unique permeation and pharmacological properties.
CryoEM structure of mGlu6 captures receptor activation prior to G protein coupling.
The metabotropic glutamate receptor 6 (mGlu6) is essential for synaptic communication of rod photoreceptors, and mutations in mGlu6 lead to a blinding disorder. However, its structural organization remains unknown. Here, we present the structure of agonist-bound mGlu6, revealing an asymmetric dimer arrangement in the absence of a G protein. This indicates that agonist binding alone can induce the homodimeric receptor asymmetry in metabotropic glutamate receptors and structurally prime mGlu6 for activation by pre-organizing the transmembrane domain dimer interface for G protein binding. The structure also identifies noncanonical interactions between the cysteine-rich domain and extracellular loop 2, forming a unique interface that likely stabilizes the activation state. Mutational analyses of this interface reveal its role in maintaining rapid Gαo activation and surface targeting. The structure also permits mechanistic investigation of congenital stationary night blindness and reveals diverse effects of pathogenic mutations on surface trafficking, Gαo coupling, and activation dynamics, including unexpected gain-of-function. These results provide critical insight into the intermediate asymmetric structure of mGlu6 and offer a molecular framework for understanding the pathogenesis of inherited retinal disorders.
EGFLAM Pathogenic Variants and Congenital Stationary Night Blindness.
Congenital stationary night blindness (CSNB) is a clinically and genetically heterogeneous inherited retinal disorder (IRD), and in many complete CSNB (cCSNB) cases, the underlying genetic cause remains unknown. Uncovering the genetic defects of IRDs helps to refine diagnostic methods and supports the development of specific therapeutic approaches. To describe the phenotype and the underlying gene defect in patients with cCSNB from 2 unrelated families. This retrospective case series was conducted from January 2023 to July 2025. Data for 3 patients from cohorts of genetically unsolved IRD cases in France (n = 140 for CSNB) and the Netherlands (n = 2730 for IRD) were analyzed clinically and genetically. Complete ocular examination, including multimodal retinal imaging and full-field electroretinography (ffERG) incorporating the International Society for Clinical Electrophysiology of Vision standards and multimodal retinal imaging, were performed. Gene defects were identified by genome sequencing (GS) and exome sequencing (ES). The main outcome was a gene defect, EGFLAM, underlying cCSNB. Measures included phenotyping, GS, ES, Sanger sequencing, and cosegregation analysis. The series included 3 patients from 2 unrelated families of Moroccan ancestry showing high myopia, reduced visual acuity, and night blindness. Retinal imaging depicted myopic changes. ffERG revealed electronegative Schubert-Bornschein configuration in keeping with cCSNB with ON-bipolar cell dysfunction. Patients were lacking pathogenic variants in known genes implicated in IRDs, including CSNB. Two different homozygous pathogenic variants, c.1563_1566del, p.(Val522Glufs*18) and c.1795C>T, p.(Arg599*) in EGFLAM were identified by ES and GS. The corresponding protein is localized in the outer plexiform layer and important for ON-bipolar cell signaling in the retina. This case series reports on a gene defect in EGFLAM implicated in human cCSNB. Clinicians should be aware about this association and consider including EGFLAM in diagnostic gene panels for IRDs. This discovery may lead to faster and more accurate diagnosis of cCSNB and genetic counseling, as well as a pathway for developing therapies.
A New Gene for Congenital Stationary Night Blindness.
G-protein activation of the dark-state conformation of the visual G protein-coupled receptor rhodopsin by releasing critical structural constraints.
G protein-coupled receptors (GPCRs) operate through the binding and activation of heterotrimeric G proteins. Ligand interaction drives the receptor's transition from an inactive to an active state, ultimately triggering G-protein activation and downstream signal transduction. The visual GPCR rhodopsin contains the chromophore 11-cis-retinal, which is covalently bound and functions as an inverse agonist, maintaining very low basal activity in the absence of light. Disruption of this basal receptor activity can lead to physiological consequences associated with retinal diseases such as congenital stationary night blindness and retinitis pigmentosa. Here, we describe a functional dark-state rhodopsin generated through engineered double and triple mutations at three well-defined structural microswitches that regulate the conformational stability of the dark ground-state: i) T942.61I, located near the protonated Schiff base linkage environment and linked to congenital stationary night blindness; ii) M2576.40Y positioned close to the tyrosine cluster (Y2235.58 and Y3067.53 of the NPxxY motif); and iii) E1343.49 within the conserved (D/E)RY motif, which participates in the so-called ionic lock involving R1353.50 and E2476.30. Characterization of these mutant rhodopsins provides additional insights into the structural basis of the inactive-to-active conformational transition in important functional domains and emphasizes rhodopsin conformational flexibility.
Publicações recentes
Autosomal dominant Riggs-type congenital stationary night blindness with fundus sheen and retinal atrophy due to a novel GNAT1 p.Gln200Arg variant.
Cryo-EM structure of TRPM1 reveals a non-canonical architecture with an inverted transmembrane domain.
CryoEM structure of mGlu6 captures receptor activation prior to G protein coupling.
G-protein activation of the dark-state conformation of the visual G protein-coupled receptor rhodopsin by releasing critical structural constraints.
Identification and functional validation of a novel disease-causing variant in the noncoding region of NYX.
📚 EuropePMC295 artigos no totalmostrando 197
Cryo-EM structure of TRPM1 reveals a non-canonical architecture with an inverted transmembrane domain.
Nature communicationsCryoEM structure of mGlu6 captures receptor activation prior to G protein coupling.
Nature communicationsG-protein activation of the dark-state conformation of the visual G protein-coupled receptor rhodopsin by releasing critical structural constraints.
Communications biologyIdentification and functional validation of a novel disease-causing variant in the noncoding region of NYX.
Acta ophthalmologicaAre There Night Driving Restrictions for Patients with Incomplete Congenital Stationary Night Blindness?
Klinische Monatsblatter fur AugenheilkundeEGFLAM Pathogenic Variants and Congenital Stationary Night Blindness.
JAMA ophthalmologyA New Gene for Congenital Stationary Night Blindness.
JAMA ophthalmologyGenetic Spectrum of Negative Electroretinograms in a Predominantly Pediatric Cohort of 177 Patients.
Investigative ophthalmology & visual sciencePattern Electroretinography Signal Reconstruction in Rare Eye Diseases using Wavelet Transform.
Annual International Conference of the IEEE Engineering in Medicine and Biology Society. IEEE Engineering in Medicine and Biology Society. Annual International ConferenceMild RPE65-Associated Inherited Retinal Dystrophies: A Multimodal Clinical and Genetic Evaluation.
Translational vision science & technologyQuantitative Proteomics Identifies Potential Molecular Adaptations in Mouse Models of Congenital Stationary Night Blindness Type 2.
Molecular & cellular proteomics : MCPIncomplete congenital stationary night blindness associated with a novel variant in the CACNA1F gene.
Documenta ophthalmologica. Advances in ophthalmologyAtypical presentation of Oguchi disease with severe cystoid macular edema and compound heterozygous SAG pathogenic variants.
Ophthalmic geneticsA mechanism for pathological oscillations in mouse retinal ganglion cells in a model of night blindness.
The Journal of general physiologyThe physiology of dark adaptation: Progress and future directions.
Progress in retinal and eye researchDomain-specific functions of LRIT3 in synaptic assembly and retinal signal transmission.
bioRxiv : the preprint server for biologyNatural course of refractive errors in early onset inherited retinal diseases.
Eye (London, England)Novel Grm6 Variant in a no b-wave (nob) Mouse Model: Phenotype Characterization and Gene Therapy.
Investigative ophthalmology & visual scienceA Practical Introduction to Wavelet Analysis in Electroretinography.
medRxiv : the preprint server for health sciencesGene augmentation therapy treats mature mice with complete congenital stationary night blindness (cCSNB), improving retinal function and visual acuity.
bioRxiv : the preprint server for biologyNovel structural variant in CACNA1F causing congenital stationary night blindness identified with whole genome sequencing.
Ophthalmic geneticsWhole exome sequencing reveals pathogenic variants in CNGA3, CACNA1F, and RPGRIP1 in consanguineous Pakistani families with diverse retinal phenotypes.
PloS oneCongenital Stationary Night Blindness.
Advances in experimental medicine and biologyElectronegative electroretinograms in two siblings with bradyopsia, an underdiagnosed pediatric inherited retinal disorder.
Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and StrabismusTrends and Disparities in the Incidence and Prevalence of Inherited Retinal Diseases in the United States.
American journal of ophthalmologyDivergent mechanisms of neural adaptation and instability in the mammalian retina.
Current biology : CBCharacterisation and prevalence of inherited retinal diseases in the Finnish population reveals enrichment of population-specific phenotypes and causative variants.
The British journal of ophthalmologyGenetic analysis of congenital stationary night blindness and Oguchi disease in an Indian cohort.
Acta ophthalmologicaReview of Four Refined Clinical Entities in Hereditary Retinal Disorders from Japan.
International journal of molecular sciencesDark Adaptometry as a Diagnostic Tool in Retinal Diseases: Mechanisms and Clinical Utility.
Journal of clinical medicineUnderstanding the phenotype of genetically associated electronegative ERG retinopathies: comparing the full-field ERG b:a ratio.
Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle OphthalmologieȦland Island eye disease in two patients harboring novel CACNA1F variants.
Ophthalmic geneticsA New Phenotypic Expression in a Patient With a Mutation in the CACNA1F Gene.
CureusGene therapy shines light on congenital stationary night blindness for future cures.
Journal of translational medicineGenetic testing as a tool for diagnosis of congenital stationary night blindness (CSNB) in white spotted breeds in Poland.
Journal of equine veterinary scienceCongenital Stationary Night Blindness (CSNB)-Case Reports and Review of Current Knowledge.
Journal of clinical medicineThe negative off-response driven by M-cone and depolarizing bipolar cell in the rat electroretinogram.
Scientific reportsDefective glycosylation and ELFN1 binding of mGluR6 congenital stationary night blindness mutants.
Life science allianceNovel CACNA1F pathogenic variant in pediatric incomplete X-linked CSNB: integrating portable ERG and genetic analysis.
Documenta ophthalmologica. Advances in ophthalmologyNatural Course of Refractive Error in Congenital Stationary Night Blindness: Implications for Myopia Treatment.
Investigative ophthalmology & visual scienceInfantile Nystagmus Syndrome-Associated Inherited Retinal Diseases: Perspectives from Gene Therapy Clinical Trials.
Life (Basel, Switzerland)Optic Neuropathy AFG3L2 Related in a Patient Affected by Congenital Stationary Night Blindness.
Case reports in ophthalmological medicineA non-conducting role of the Cav1.4 Ca2+ channel drives homeostatic plasticity at the cone photoreceptor synapse.
eLifeCryo-EM structure of human class C orphan GPCR GPR179 involved in visual processing.
Nature communicationsLoss of ON-Pathway Function in Mice Lacking Lrit3 Decreases Recovery From Lens-Induced Myopia.
Investigative ophthalmology & visual scienceDifferential pathogenetic mechanisms of mutations in helix 2 and helix 6 of rhodopsin.
International journal of biological macromoleculesA novel homozygous nonsense variant in CABP4 causing stationary cone/rod synaptic dysfunction.
Ophthalmic geneticsAnti-TRPM1 autoantibody-positive unilateral melanoma associated retinopathy (MAR) triggered by immunotherapy recapitulates functional and structural details of TRPM1-associated congenital stationary night blindness.
American journal of ophthalmology case reportsCharacterising the refractive error in paediatric patients with congenital stationary night blindness: a multicentre study.
The British journal of ophthalmologyCross-species single-cell landscapes identify the pathogenic gene characteristics of inherited retinal diseases.
Frontiers in geneticsGenetic Characterization of 191 Probands with Inherited Retinal Dystrophy by Targeted NGS Analysis.
GenesCharacterizing Retinal Sensitivity and Structure in Congenital Stationary Night Blindness: A Combined Microperimetry and OCT Study.
Investigative ophthalmology & visual scienceNationwide Prevalence of Inherited Retinal Diseases in the Israeli Population.
JAMA ophthalmologyDark continuous noise from mutant G90D-rhodopsin predominantly underlies congenital stationary night blindness.
Proceedings of the National Academy of Sciences of the United States of AmericaDiagnosis of Incomplete Congenital Stationary Night Blindness in a 2-year-old boy.
Klinische Monatsblatter fur AugenheilkundeA Case of Congenital Stationary Night Blindness in a Healthy Female Infant: Emphasis on Electroretinography.
Klinische Monatsblatter fur AugenheilkundeCongenital Stationary Night Blindness: Structure, Function and Genotype-Phenotype Correlations in a Cohort of 122 Patients.
Ophthalmology. RetinaRPE65-Associated Retinal Dystrophies: Phenotypes and Treatment Effects with Voretigene Neparvovec.
Klinische Monatsblatter fur AugenheilkundeAland Island Eye Disease with Retinoschisis in the Clinical Spectrum of CACNA1F-Associated Retinopathy-A Case Report.
International journal of molecular sciencesClinical and genetic studies for a cohort of patients with congenital stationary night blindness.
Orphanet journal of rare diseasesCompound heterozygous mutations in GRM6 causing complete Schubert-Bornschein type congenital stationary night blindness.
HeliyonA Drosophila Model Reveals the Potential Role for mtt in Retinal Disease.
International journal of molecular sciencesUnusual OCT findings in a patient with CABP4-associated cone-rod synaptic disorder.
Documenta ophthalmologica. Advances in ophthalmologyCACNA1F-related synaptic dysfunction: challenges diagnosing congenital stationary night blindness presenting without night blindness.
Canadian journal of ophthalmology. Journal canadien d'ophtalmologieGrk1 Missense Mutations in Type II Oguchi Disease: A Literature Review.
Annals of biomedical researchHIGH MYOPIA IS COMMON IN PATIENTS WITH X-LINKED RETINOPATHIES: Myopic Maculopathy Analysis.
Retina (Philadelphia, Pa.)Oguchi's disease - Clinical image.
Oman journal of ophthalmologyFundus Albipunctatus Associated with Biallelic LRAT Gene Mutation: A Case Report with Long-Term Follow-Up.
Journal of clinical medicineThe rod synapse in aging wildtype and Dscaml1 mutant mice.
PloS oneA common cause for nystagmus in different congenital stationary night blindness mouse models.
The Journal of physiologyThe Value of Electroretinography in Identifying Candidate Genes for Inherited Retinal Dystrophies: A Diagnostic Guide.
Diagnostics (Basel, Switzerland)Additional evidence supports GRM6 p.Thr178Met as a cause of congenital stationary night blindness in three horse breeds.
Veterinary ophthalmologyGene Therapy in Hereditary Retinal Dystrophies: The Usefulness of Diagnostic Tools in Candidate Patient Selections.
International journal of molecular sciencesA retinal origin of nystagmus-a perspective.
Frontiers in ophthalmologyGenetic and Clinical Profile of Retinopathies Due to Disease-Causing Variants in Leber Congenital Amaurosis (LCA)-Associated Genes in a Large German Cohort.
International journal of molecular sciencesExtended functional rescue following AAV gene therapy in a canine model of LRIT3-congenital stationary night blindness.
Vision researchCanine and Feline Models of Inherited Retinal Diseases.
Cold Spring Harbor perspectives in medicineCav1.4 congenital stationary night blindness is associated with an increased rate of proteasomal degradation.
Frontiers in cell and developmental biologyMouse all-cone retina models of Cav1.4 synaptopathy.
Frontiers in molecular neuroscienceLRIT3 expression in cone photoreceptors restores post-synaptic bipolar cell signalplex assembly and partial function in Lrit3 -/- mice.
iScienceCharacterization of two pathological gating-charge substitutions in Cav1.4 L-type calcium channels.
Channels (Austin, Tex.)Whale shark rhodopsin adapted to deep-sea lifestyle by a substitution associated with human disease.
Proceedings of the National Academy of Sciences of the United States of America[Nystagmus in Children - a Survey].
Klinische Monatsblatter fur AugenheilkundeShedding light on myopia by studying complete congenital stationary night blindness.
Progress in retinal and eye researchCorrelation between the Serum Concentration of Vitamin A and Disease Severity in Patients Carrying p.G90D in RHO, the Most Frequent Gene Associated with Dominant Retinitis Pigmentosa: Implications for Therapy with Vitamin A.
International journal of molecular sciencesMice Lacking Gpr179 with Complete Congenital Stationary Night Blindness Are a Good Model for Myopia.
International journal of molecular sciencesUpward saccadic intrusions as the presenting feature for incomplete congenital stationary night blindness.
Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and StrabismusCongenital Stationary Night Blindness: Clinical and Genetic Features.
International journal of molecular sciencesOptic nerve involvement in CACNA1F-related disease: observations from a multicentric case series.
Ophthalmic geneticsGenetic analysis and clinical features of three Chinese patients with Oguchi disease.
Documenta ophthalmologica. Advances in ophthalmologyIdentification of a novel large multigene deletion and a frameshift indel in PDE6B as the underlying cause of early-onset recessive rod-cone degeneration.
Cold Spring Harbor molecular case studiesDelivery strategies for CRISPR/Cas genome editing tool for retinal dystrophies: challenges and opportunities.
Asian journal of pharmaceutical sciencesAssociation of Missense Variants in VSX2 With a Peculiar Form of Congenital Stationary Night Blindness Affecting All Bipolar Cells.
JAMA ophthalmologyMultimodal imaging in Schubert-Bornschein congenital stationary night blindness.
Ophthalmic geneticsAssessing the Pathogenicity of In-Frame CACNA1F Indel Variants Using Structural Modeling.
The Journal of molecular diagnostics : JMDInvolvement of transient receptor potential channels in ocular diseases: a narrative review.
Annals of translational medicineA clinical and electrophysiological case study of a child with a novel frame shift mutation in the CACNA1F and missense variation of RIMS1 genes.
Documenta ophthalmologica. Advances in ophthalmologyUnilateral cataract and congenital stationary night blindness in a child with novel variants in TRPM1.
Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and StrabismusMolecular basis for variations in the sensitivity of pathogenic rhodopsin variants to 9-cis-retinal.
The Journal of biological chemistryTwo novel CACNA1F gene mutations cause two different phenotypes: Aland Eye Disease and incomplete Congenital Stationary Night Blindness.
Experimental eye researchPhenotypes and genotypes underlying paradoxical pupillary reaction in children.
Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and StrabismusClinical and genetic findings in TRPM1-related congenital stationary night blindness.
Acta ophthalmologicaMutation analysis reveals novel and known mutations in SAG gene in first two Egyptian families with Oguchi disease.
BMC ophthalmologyIdentification of autosomal recessive novel genes and retinal phenotypes in members of the solute carrier (SLC) superfamily.
Genetics in medicine : official journal of the American College of Medical GeneticsSLC24A1-Associated Congenital Stationary Night Blindness in a Woman With an Abnormal Fundus.
JAMA ophthalmologyA novel missense creatine mutant of CaBP4, c.464G>A (p.G155D), associated with autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE), reduces the expression of CaBP4.
Translational pediatricsTargeting ON-bipolar cells by AAV gene therapy stably reverses LRIT3-congenital stationary night blindness.
Proceedings of the National Academy of Sciences of the United States of AmericaElectronegative electroretinogram in the modern multimodal imaging era.
Clinical & experimental ophthalmologyPosterior staphyloma with congenital stationary night blindness.
Journal francais d'ophtalmologieGenetic Analysis of Consanguineous Pakistani Families with Congenital Stationary Night Blindness.
Ophthalmic researchComments on 'Whole-genome sequencing identifies missense mutation in GRM6 as the likely cause of congenital stationary night blindness in a Tennessee Walking Horse'.
Equine veterinary journalSubstantial restoration of night vision in adult mice with congenital stationary night blindness.
Molecular therapy. Methods & clinical developmentNon-syndromic inherited retinal diseases in Poland: Genes, mutations, and phenotypes.
Molecular visionThe role of voltage-gated ion channels in visual function and disease in mammalian photoreceptors.
Pflugers Archiv : European journal of physiologyCav1.4 dysfunction and congenital stationary night blindness type 2.
Pflugers Archiv : European journal of physiologyNYX-related Congenital Stationary Night Blindness in Two Siblings due to Probable Maternal Germline Mosaicism.
Ophthalmic geneticsTwo novel compound heterozygous SAG mutations in an Italian patient with Oguchi disease: A genetic and multimodal retinal imaging study.
European journal of ophthalmologyNegative electroretinograms: genetic and acquired causes, diagnostic approaches and physiological insights.
Eye (London, England)Clinical and Genetic Characteristics of Korean Congenital Stationary Night Blindness Patients.
GenesOguchi's disease: two cases and literature review.
The Journal of international medical researchFundus albipunctatus photoreceptor microstructure revealed using adaptive optics scanning light ophthalmoscopy.
American journal of ophthalmology case reportsRetinal imaging in inherited retinal diseases.
Annals of eye scienceA New Mouse Model for Complete Congenital Stationary Night Blindness Due to Gpr179 Deficiency.
International journal of molecular sciencesComplete congenital stationary night blindness associated with a novel NYX variant (p.Asn216Lys) in middle-aged and older adult patients.
Ophthalmic geneticsPanel-based genetic testing for inherited retinal disease screening 176 genes.
Molecular genetics & genomic medicineRestoration of mGluR6 Localization Following AAV-Mediated Delivery in a Mouse Model of Congenital Stationary Night Blindness.
Investigative ophthalmology & visual scienceStructural aspects of rod opsin and their implication in genetic diseases.
Pflugers Archiv : European journal of physiologyWhole-exome sequencing in 168 Korean patients with inherited retinal degeneration.
BMC medical genomicsCongenital stationary night blindness in a patient with mild learning disability due to a compound heterozygous microdeletion of 15q13 and a missense mutation in TRPM1.
Ophthalmic geneticsStationary and Progressive Phenotypes Caused by the p.G90D Mutation in Rhodopsin Gene.
International journal of molecular sciencesOptic Atrophy and Inner Retinal Thinning in CACNA1F-related Congenital Stationary Night Blindness.
Genes[Decreased visual acuity as presenting sign of a late case of congenital stationary night blindness: The role of ERG].
Journal francais d'ophtalmologieA Review of Genetic and Physiological Disease Mechanisms Associated With Cav1 Channels: Implications for Incomplete Congenital Stationary Night Blindness Treatment.
Frontiers in geneticsFunction of cone and cone-related pathways in CaV1.4 IT mice.
Scientific reportsA Novel Splice-Site Variant in CACNA1F Causes a Phenotype Synonymous with Åland Island Eye Disease and Incomplete Congenital Stationary Night Blindness.
GenesCongenital stationary night blindness: an update and review of the disease spectrum in Saudi Arabia.
Acta ophthalmologicaNew variants and in silico analyses in GRK1 associated Oguchi disease.
Human mutationTransgenic Expression of Cacna1f Rescues Vision and Retinal Morphology in a Mouse Model of Congenital Stationary Night Blindness 2A (CSNB2A).
Translational vision science & technologyGUCY2D mutations in retinal guanylyl cyclase 1 provide biochemical reasons for dominant cone-rod dystrophy but not for stationary night blindness.
The Journal of biological chemistryNovel biallelic TRPM1 variants in an elderly patient with complete congenital stationary night blindness.
Documenta ophthalmologica. Advances in ophthalmologyFunctional impact of a congenital stationary night blindness type 2 mutation depends on subunit composition of Cav1.4 Ca2+ channels.
The Journal of biological chemistrySensing through Non-Sensing Ocular Ion Channels.
International journal of molecular sciencesA Homozygote Mutation in S-Antigen Visual Arrestin SAG Gene in an Iranian Patient with Oguchi Type One: A Case Report.
Iranian journal of public healthLIM-Homeodomain Transcription Factor LHX4 Is Required for the Differentiation of Retinal Rod Bipolar Cells and OFF-Cone Bipolar Subtypes.
Cell reportsPhenotype Driven Analysis of Whole Genome Sequencing Identifies Deep Intronic Variants that Cause Retinal Dystrophies by Aberrant Exonization.
Investigative ophthalmology & visual scienceElectronegative Electroretinograms in the United Arab Emirates.
Middle East African journal of ophthalmologyThe X-linked retinopathies: Physiological insights, pathogenic mechanisms, phenotypic features and novel therapies.
Progress in retinal and eye researchDiagnosis of an X-linked type 2 congenital stationary night blindness using electroretinography and CACNA1F sequencing.
Archivos de la Sociedad Espanola de OftalmologiaGenetics of Equine Ocular Disease.
The Veterinary clinics of North America. Equine practiceWhole-genome sequencing identifies missense mutation in GRM6 as the likely cause of congenital stationary night blindness in a Tennessee Walking Horse.
Equine veterinary journalNext generation sequencing using phenotype-based panels for genetic testing in inherited retinal diseases.
Ophthalmic geneticsDifferential adaptations in rod outer segment disc membranes in different models of congenital stationary night blindness.
Biochimica et biophysica acta. BiomembranesLoss of Function of RIMS2 Causes a Syndromic Congenital Cone-Rod Synaptic Disease with Neurodevelopmental and Pancreatic Involvement.
American journal of human geneticsA Conserved Proline Hinge Mediates Helix Dynamics and Activation of Rhodopsin.
Structure (London, England : 1993)Using an integrative machine learning approach utilising homology modelling to clinically interpret genetic variants: CACNA1F as an exemplar.
European journal of human genetics : EJHGLarge Animal Models of Inherited Retinal Degenerations: A Review.
CellsTargeting molecular pathways for the treatment of inherited retinal degeneration.
Neural regeneration researchCongenital Stationary Night Blindness due to Novel TRPM1 Gene Mutations in a Korean Patient.
Korean journal of ophthalmology : KJOWide-field true-colour imaging and clinical characterization of a novel GRK1 mutation in Oguchi disease.
Documenta ophthalmologica. Advances in ophthalmologyRing analysis of multifocal oscillatory potentials (mfOPs) in cCSNB suggests near-normal ON-OFF pathways at the fovea only.
Documenta ophthalmologica. Advances in ophthalmologyComprehensive Geno- and Phenotyping in a Complex Pedigree Including Four Different Inherited Retinal Dystrophies.
GenesRetinal degeneration in mice expressing the constitutively active G90D rhodopsin mutant.
Human molecular geneticsA founder RDH5 splice site mutation leads to retinitis punctata albescens in two inbred Pakistani kindreds.
Ophthalmic geneticsLong-term follow-up of retinal function and structure in TRPM1-associated complete congenital stationary night blindness.
Molecular visionElectroretinographic abnormalities associated with pregabalin: a case report.
Documenta ophthalmologica. Advances in ophthalmologyNystagmus with pendular low amplitude, high frequency components (PLAHF) in association with retinal disease.
StrabismusGenetic Deciphering of Early-Onset and Severe Retinal Dystrophy Associated with Sensorineural Hearing Loss.
Advances in experimental medicine and biologyEarly onset retinal dystrophies: clinical clues to diagnosis for pediatricians.
Italian journal of pediatricsNovel frameshift mutation in NYX gene in a Russian family with complete congenital stationary night blindness.
Ophthalmic geneticsNovel homozygous in-frame deletion of GNAT1 gene causes golden appearance of fundus and reduced scotopic ERGs similar to that in Oguchi disease in Japanese family.
Ophthalmic geneticsPseudodominant inheritance of autosomal recessive congenital stationary night blindness in one family with three co-segregating deleterious GRM6 variants identified by next-generation sequencing.
Molecular genetics & genomic medicineAn Ashkenazi Jewish founder mutation in CACNA1F causes retinal phenotype in both hemizygous males and heterozygous female carriers.
Ophthalmic geneticsA founder deletion in the TRPM1 gene associated with congenital stationary night blindness and myopia is highly prevalent in Ashkenazi Jews.
Human genome variationExtracting the ON and OFF contributions to the full-field photopic flash electroretinogram using summed growth curves.
Experimental eye researchMolecular mechanisms underlying selective synapse formation of vertebrate retinal photoreceptor cells.
Cellular and molecular life sciences : CMLSCoexistence of GNAT1 and ABCA4 variants associated with Nougaret-type congenital stationary night blindness and childhood-onset cone-rod dystrophy.
Documenta ophthalmologica. Advances in ophthalmologyGenome-wide association study and whole-genome sequencing identify a deletion in LRIT3 associated with canine congenital stationary night blindness.
Scientific reportsNystagmus in patients with congenital stationary night blindness (CSNB) originates from synchronously firing retinal ganglion cells.
PLoS biologyTRPM1 Mutations are the Most Common Cause of Autosomal Recessive Congenital Stationary Night Blindness (CSNB) in the Palestinian and Israeli Populations.
Scientific reportsPresynaptic Expression of LRIT3 Transsynaptically Organizes the Postsynaptic Glutamate Signaling Complex Containing TRPM1.
Cell reportsGenetic Mutation Profiles in Korean Patients with Inherited Retinal Diseases.
Journal of Korean medical scienceIn vivo electroretinographic differentiation of rod, short-wavelength and long/medium-wavelength cone responses in dogs using silent substitution stimuli.
Experimental eye researchReading Individual Words Within Sentences in Infantile Nystagmus.
Investigative ophthalmology & visual scienceIdentification of a novel GRM6 mutation in a previously described consanguineous family with complete congenital stationary night blindness.
Ophthalmic geneticsDisinhibition of intrinsic photosensitive retinal ganglion cells in patients with X-linked congenital stationary night blindness.
Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie[Multimodal diagnostic of CSNB1 with NYX gene mutation].
Der Ophthalmologe : Zeitschrift der Deutschen Ophthalmologischen GesellschaftA Mixture of U.S. Food and Drug Administration-Approved Monoaminergic Drugs Protects the Retina From Light Damage in Diverse Models of Night Blindness.
Investigative ophthalmology & visual scienceMizuo-Nakamura phenomenon in an Indian male.
Clinical case reportsWhere are the missing gene defects in inherited retinal disorders? Intronic and synonymous variants contribute at least to 4% of CACNA1F-mediated inherited retinal disorders.
Human mutationThree cases of acute-onset bilateral photophobia.
Japanese journal of ophthalmologyMacular sensitivity in patients with congenital stationary night-blindness.
The British journal of ophthalmologyPhotoreceptor degeneration in a new Cacna1f mutant mouse model.
Experimental eye researchSplicing of an automodulatory domain in Cav1.4 Ca2+ channels confers distinct regulation by calmodulin.
The Journal of general physiologyNovel truncating mutation in CACNA1F in a young male patient diagnosed with optic atrophy.
Ophthalmic geneticsUnexpected Genetic Cause in Two Female Siblings with High Myopia and Reduced Visual Acuity.
BioMed research internationalCongenital stationary night blindness associated with morning glory disc malformation: a novel hemizygous mutation in CACNA1F.
Ophthalmic geneticsPhenotypic characterization of complete CSNB in the inbred research beagle: how common is CSNB in research and companion dogs?
Documenta ophthalmologica. Advances in ophthalmologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Cegueira noturna estacionária congênita.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Cegueira noturna estacionária congênita
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Cryo-EM structure of TRPM1 reveals a non-canonical architecture with an inverted transmembrane domain.
- CryoEM structure of mGlu6 captures receptor activation prior to G protein coupling.
- EGFLAM Pathogenic Variants and Congenital Stationary Night Blindness.
- A New Gene for Congenital Stationary Night Blindness.
- G-protein activation of the dark-state conformation of the visual G protein-coupled receptor rhodopsin by releasing critical structural constraints.
- Autosomal dominant Riggs-type congenital stationary night blindness with fundus sheen and retinal atrophy due to a novel GNAT1 p.Gln200Arg variant.
- Identification and functional validation of a novel disease-causing variant in the noncoding region of NYX.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:215(Orphanet)
- MONDO:0016293(MONDO)
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q18553290(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Cegueira noturna estacionária congênita
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov