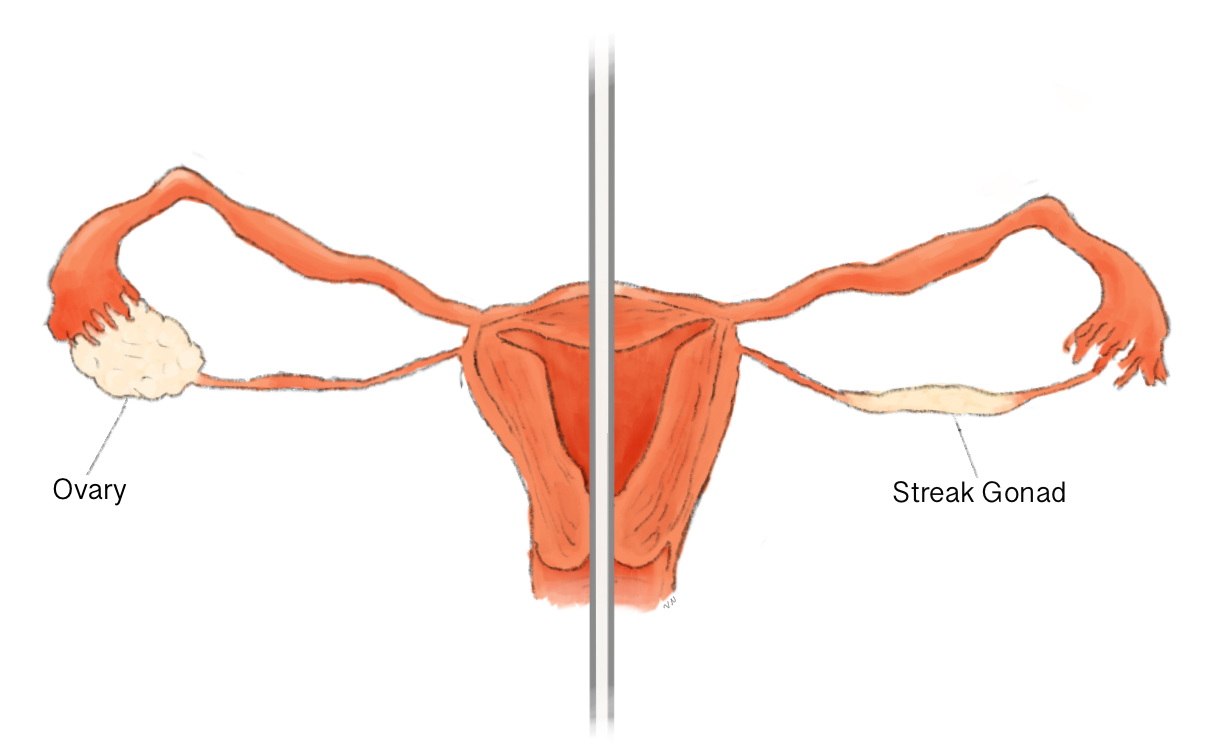
A disgenesia gonadal 46,XX (DG 46,XX) é um problema primário nos ovários que leva à falência ovariana prematura (FOP), também conhecida como menopausa precoce, em mulheres com cromossomos 46,XX que, de outra forma, seriam normais. Isso acontece porque os ovários não se desenvolvem ou porque não respondem aos hormônios (gonadotrofinas) que deveriam estimulá-los.
Introdução
O que você precisa saber de cara
A disgenesia gonadal 46,XX (DG 46,XX) é um problema primário nos ovários que leva à falência ovariana prematura (FOP), também conhecida como menopausa precoce, em mulheres com cromossomos 46,XX que, de outra forma, seriam normais. Isso acontece porque os ovários não se desenvolvem ou porque não respondem aos hormônios (gonadotrofinas) que deveriam estimulá-los.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 23 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 63 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
16 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive, Not applicable, X-linked recessive.
Mitochondrion
Combined oxidative phosphorylation deficiency 5
A mitochondrial disease resulting in severe metabolic acidosis, edema, hypertrophic cardiomyopathy, tubulopathy, and hypotonia.
Nuclear hormone receptor. Binds estrogens with an affinity similar to that of ESR1/ER-alpha, and activates expression of reporter genes containing estrogen response elements (ERE) in an estrogen-dependent manner (PubMed:20074560) Lacks ligand binding ability and has no or only very low ERE binding activity resulting in the loss of ligand-dependent transactivation ability
Nucleus
Ovarian dysgenesis 8
An autosomal dominant form of ovarian dysgenesis, a disorder characterized by lack of spontaneous pubertal development, primary amenorrhea, uterine hypoplasia, and hypergonadotropic hypogonadism as a result of streak gonads.
Transcription regulator of both male and female germline differentiation. Suppresses genes involved in spermatogonial stem cells maintenance, and induces genes important for spermatogonial differentiation. Coordinates oocyte differentiation without affecting meiosis I (By similarity)
CytoplasmNucleus
Spermatogenic failure 32
An autosomal dominant infertility disorder caused by spermatogenesis defects that result in non-obstructive azoospermia.
Ligand for members of the frizzled family of seven transmembrane receptors (Probable). Plays an important role in the embryonic development of the urogenital tract and the lung (PubMed:15317892, PubMed:16959810, PubMed:18179883, PubMed:18182450). Required for normal mesenchyme to epithelium transition during embryonic kidney development. Required for the formation of early epithelial renal vesicles during kidney development (By similarity). Required for normal formation of the Mullerian duct in
Secreted, extracellular space, extracellular matrix
46,XX sex reversal with dysgenesis of kidneys, adrenals, and lungs
A disease characterized by the association of female-to-male sex reversal with dysgenesis of kidneys, adrenals, and lungs.
DNA-binding protein involved in homologous recombination that acts by recruiting the MCM8-MCM9 helicase complex to sites of DNA damage to promote DNA repair synthesis (PubMed:31467087). A C-terminal region including the OB-fold stimulates the helicase activity of MCM8-MCM9 probably by altering its conformation (PubMed:37535404)
NucleusChromosome
Ovarian dysgenesis 11
An autosomal recessive form of ovarian dysgenesis, a disorder characterized by lack of spontaneous pubertal development, primary amenorrhea, uterine hypoplasia, and hypergonadotropic hypogonadism as a result of streak gonads.
DNA-dependent RNA polymerase catalyzes the transcription of DNA into RNA using the four ribonucleoside triphosphates as substrates (PubMed:20413673, PubMed:33558764, PubMed:34675218). Specific peripheric component of RNA polymerase III (Pol III) which synthesizes small non-coding RNAs including 5S rRNA, snRNAs, tRNAs and miRNAs from at least 500 distinct genomic loci. With CRCP/RPC9 forms a mobile stalk that protrudes from Pol III core and functions primarily in transcription initiation (By simi
Nucleus
G protein-coupled receptor for follitropin, the follicle-stimulating hormone (PubMed:11847099, PubMed:24058690, PubMed:24692546). Through cAMP production activates the downstream PI3K-AKT and ERK1/ERK2 signaling pathways (PubMed:24058690)
Cell membrane
Ovarian dysgenesis 1
An autosomal recessive disease characterized by primary amenorrhea, variable development of secondary sex characteristics, poorly developed streak ovaries, and high serum levels of follicle-stimulating hormone (FSH) and luteinizing hormone (LH).
Plays a role in the nuclear pore complex (NPC) assembly and/or maintenance (PubMed:12552102, PubMed:15229283, PubMed:30179222). Required for the assembly of peripheral proteins into the NPC (PubMed:12552102, PubMed:15229283). May anchor NUP62 to the NPC (PubMed:15229283). Involved in nephrogenesis (PubMed:30179222)
Nucleus membraneNucleus, nuclear pore complexChromosome, centromere, kinetochore
Nephrotic syndrome 11
A form of nephrotic syndrome, a renal disease clinically characterized by severe proteinuria, resulting in complications such as hypoalbuminemia, hyperlipidemia and edema. Kidney biopsies show non-specific histologic changes such as focal segmental glomerulosclerosis and diffuse mesangial proliferation. Some affected individuals have an inherited steroid-resistant form and progress to end-stage renal failure. NPHS11 is an autosomal recessive, steroid-resistant and progressive form with onset in the first decade of life.
Transcriptional activator. Essential for sexual differentiation and formation of the primary steroidogenic tissues (PubMed:27378692). Binds to the Ad4 site found in the promoter region of steroidogenic P450 genes such as CYP11A, CYP11B and CYP21B. Also regulates the AMH/Muellerian inhibiting substance gene as well as the AHCH and STAR genes. 5'-YCAAGGYC-3' and 5'-RRAGGTCA-3' are the consensus sequences for the recognition by NR5A1 (PubMed:27378692). The SFPQ-NONO-NR5A1 complex binds to the CYP17
Nucleus
46,XY sex reversal 3
A condition characterized by male-to-female sex reversal in the presence of a normal 46,XY karyotype.
Plays a role in DNA double-strand break (DBS) repair via homologous recombination (HR). Serves as a scaffolding protein that helps to promote the recruitment of DNA-processing enzymes like the helicase BLM and recombinase RAD51 to site of DNA damage, and hence contributes to maintain genomic integrity
Nucleus
Ovarian dysgenesis 9
An autosomal recessive form of ovarian dysgenesis, a disorder characterized by lack of spontaneous pubertal development, primary amenorrhea, uterine hypoplasia, and hypergonadotropic hypogonadism as a result of streak gonads.
Involved in meiotic recombination. Required for reciprocal recombination and proper segregation of homologous chromosomes at meiosis
Chromosome
Spermatogenic failure 2
An autosomal recessive disorder characterized by male infertility due to non-obstructive azoospermia. Testicular histopathology reveals no round spermatids or spermatozoa in the seminiferous tubules of SPGF2 patients, consistent with meiotic arrest.
Involved in early stages of the homologous recombination repair (HRR) pathway of double-stranded DNA breaks arising during DNA replication or induced by DNA-damaging agents. Required for meiotic progression, hence for fertility (PubMed:32719396, PubMed:33713115, PubMed:34402903)
Nucleus
Ovarian dysgenesis 10
An autosomal recessive form of ovarian dysgenesis, a disorder characterized by lack of spontaneous pubertal development, primary amenorrhea, uterine hypoplasia, and hypergonadotropic hypogonadism as a result of streak gonads.
Plays an important role in meiotic recombination. Stimulates DMC1-mediated strand exchange required for pairing homologous chromosomes during meiosis. The complex PSMC3IP/MND1 binds DNA, stimulates the recombinase activity of DMC1 as well as DMC1 D-loop formation from double-strand DNA. This complex stabilizes presynaptic RAD51 and DMC1 filaments formed on single strand DNA to capture double-strand DNA. This complex stimulates both synaptic and presynaptic critical steps in RAD51 and DMC1-promot
Nucleus
Ovarian dysgenesis 3
A disorder characterized by lack of spontaneous pubertal development, primary amenorrhea, uterine hypoplasia, and hypergonadotropic hypogonadism as a result of streak gonads.
May be involved in follicular development. Oocyte-specific growth/differentiation factor that stimulates folliculogenesis and granulosa cell (GC) growth
Secreted
Ovarian dysgenesis 2
A disorder characterized by lack of spontaneous pubertal development, primary amenorrhea, uterine hypoplasia, and hypergonadotropic hypogonadism as a result of streak gonads.
Transcriptional activator (By similarity). It is likely involved in the regulation of keratinocytes terminal differentiation in squamous epithelia and hair follicles (PubMed:8034748). Required for the maintenance of spermatogenesis (By similarity). It is involved in the positive regulation of oocyte maturation, probably acting through the control of BMP15 levels and regulation of AKT signaling cascade (PubMed:30010909). May also play a role in the early development of embryos (By similarity)
NucleusCytoplasmNucleus, nucleoplasm
Premature ovarian failure 16
An autosomal dominant form of premature ovarian failure, an ovarian disorder defined as the cessation of ovarian function under the age of 40 years. It is characterized by oligomenorrhea or amenorrhea, in the presence of elevated levels of serum gonadotropins and low estradiol.
Germline specific transcription factor implicated in postnatal oocyte-specific gene expression. Plays a key regulatory role in the expression of multiple oocyte-specific genes, including those that initiate folliculogenesis and those that encode the zona pellucida (ZP1, ZP2 and ZP3) required for fertilization and early embryonic survival. Essential for oocytes to survive and form primordial follicles. The persistence of FIGLA in adult females suggests that it may regulate additional pathways tha
Nucleus
Premature ovarian failure 6
An ovarian disorder defined as the cessation of ovarian function under the age of 40 years. It is characterized by oligomenorrhea or amenorrhea, in the presence of elevated levels of serum gonadotropins and low estradiol.
Variantes genéticas (ClinVar)
151 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
62 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Disgenesia gonadal 46,XX
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Mostrando amostra de 1 publicações de um total de 11
Publicações recentes
Tuberculosis treatment spills the beans on Wilson's disease and more.
Unusual combination of acute aortic dissection, Mayer-Rokitansky-Küster-Hauser syndrome, and 46,XX gonadal dysgenesis: A case report.
A rare case of 46,XX gonadal dysgenesis, Mayer-Rokitansky-Kuster-Hauser syndrome, pituitary and thyroid hypoplasia.
Misdiagnosis of associated mullerian agenesis in a female with 46, XX gonadal dysgenesis: a case report and review of literature.
Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Disgenesia gonadal 46,XX.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Disgenesia gonadal 46,XX
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Unusual combination of acute aortic dissection, Mayer-Rokitansky-Küster-Hauser syndrome, and 46,XX gonadal dysgenesis: A case report.
- WT1 Disorder.
- Tuberculosis treatment spills the beans on Wilson's disease and more.
- A rare case of 46,XX gonadal dysgenesis, Mayer-Rokitansky-Kuster-Hauser syndrome, pituitary and thyroid hypoplasia.
- Misdiagnosis of associated mullerian agenesis in a female with 46, XX gonadal dysgenesis: a case report and review of literature.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:243(Orphanet)
- MONDO:0009299(MONDO)
- GARD:5671(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q8042656(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Disgenesia gonadal 46,XX
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata