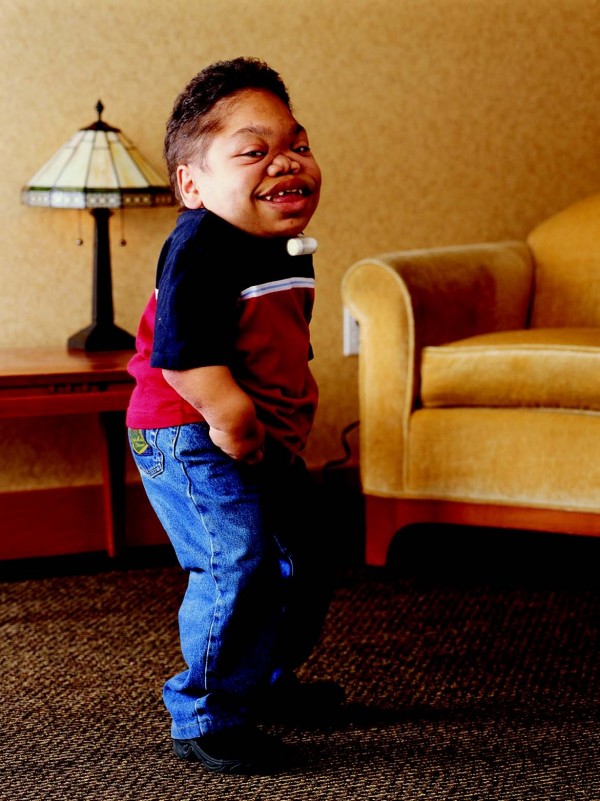
A mucolipidose III alfa/beta (MLIII alfa/beta) é uma doença lisossômica caracterizada por desaceleração progressiva da taxa de crescimento desde a primeira infância, rigidez e dor nas articulações, espessamento gradual das características faciais, atraso moderado no desenvolvimento e deficiência intelectual leve na maioria dos pacientes.
Introdução
O que você precisa saber de cara
A mucolipidose III alfa/beta (MLIII alfa/beta) é uma doença lisossômica caracterizada por desaceleração progressiva da taxa de crescimento desde a primeira infância, rigidez e dor nas articulações, espessamento gradual das características faciais, atraso moderado no desenvolvimento e deficiência intelectual leve na maioria dos pacientes.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 32 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 103 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Catalyzes the formation of mannose 6-phosphate (M6P) markers on high mannose type oligosaccharides in the Golgi apparatus. M6P residues are required to bind to the M6P receptors (MPR), which mediate the vesicular transport of lysosomal enzymes to the endosomal/prelysosomal compartment
Golgi apparatus membrane
Mucolipidosis type II
Fatal, autosomal recessive, lysosomal storage disorder characterized by severe clinical and radiologic features, peculiar fibroblast inclusions, and no excessive mucopolysacchariduria. Congenital dislocation of the hip, thoracic deformities, hernia, and hyperplastic gums are evident soon after birth.
Variantes genéticas (ClinVar)
523 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 2.737 variantes classificadas pelo ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Mucolipidose tipo III
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
When it's not juvenile idiopathic arthritis: unmasking monogenic mimickers in children monogenic mimickers of chronic arthritis.
Chronic arthritis in children is widely associated with juvenile idiopathic arthritis (JIA); however, several rare monogenic disorders may mimic its clinical presentation. Misdiagnosis can result in unnecessary immunosuppressive treatment and delay appropriate care. This study aimed to share our experience with monogenic disorders presenting as chronic arthritis and highlight their distinguishing clinical and imaging features. This retrospective cohort study included patients initially suspected of having JIA who were later diagnosed with a monogenic disorder. Clinical, laboratory, imaging, and genetic data were also collected and evaluated. Among the 25 patients, the most frequent diagnoses were progressive pseudorheumatoid dysplasia (PPRD; n = 12) and camptodactyly arthropathy-coxa vara-pericarditis (CACP) syndrome (n = 8). Other diagnoses included mucolipidosis type III gamma, primary hypertrophic osteoarthropathy (PHO), and multicentric carpotarsal osteolysis (MCTO). While all PPRD and CACP patients had biallelic pathogenic variants in CCN6 and PRG4, respectively, PHO and MCTO were associated with monoallelic mutations. Common misdiagnoses included polyarticular JIA, leading to the inappropriate use of methotrexate or biologic agents. Several monogenic disorders can mimic JIA in pediatric patients, leading to diagnostic challenges. Clinical features, such as camptodactyly, skeletal deformities, digital clubbing, median nerve neuropathy, and poor response to treatment should prompt further evaluation, including genetic testing. Increased awareness and early recognition of these conditions are crucial to avoid unnecessary immunosuppression and improve patient outcomes. • Several rare monogenic disorders can clinically mimic JIA and misdiagnosis of these monogenic mimickers may lead to inappropriate immunosuppressive treatment and delayed appropriate care. • This study presents a real-world single-center data that systematically characterizes five distinct monogenic disorders mimicking JIA. • Findings emphasize the importance of early suspicion, especially in cases with symmetrical interphalangeal involvement (PPRD), camptodactyly with hip deformity (CACP), or osteolysis with renal/facial anomalies (MCTO), to avoid unnecessary immunosuppression.
Atypical presentation of mucolipidosis type III.
Hand stiffness not only a rheumatological sign: A case of early onset mucolipidosis III-gamma with literature review.
Mucolipidosis (ML) is a rare autosomal recessive lysosomal disorder with variable onset and severity: MLII, characterized by early onset and rapid progression, and MLIII, milder with late onset and prolonged survival. ML is due to mutations in the Golgi enzyme uridine diphosphate-N-acetylglucosamine-1-phosphotransferase, whose subunits are encoded by GNPTAB and GNPTG genes. This report presents a particular case of infantile early-onset MLIII-gamma and emphasizes that articular manifestations can be a sign of a metabolic disease rather than a rheumatological or orthopedic one. A 5.7-years-old girl presented with progressive hand stiffness and joint pain, exhibiting symptoms from 6 months of age. She displayed claw-hand deformity and joint stiffness but normal growth and neurodevelopment. Biochemical testing revealed normal activities of alpha-L-iduronidase and arylsulfatase-B in leukocytes, excluding mucopolysaccharidosis I and VI, while beta-hexosaminidase and alpha-L-fucosidase activities in plasma were elevated, suggesting ML. Genetic analysis of GNPTAB and GNPTG identified two pathogenic variants in the GNPTG gene, confirming MLIII-gamma diagnosis. Despite early onset, the patient exhibited a less severe skeletal phenotype and showed mild cardiac and ocular involvement, occasionally described in classic MLIII-gamma. The natural history of MLIII remains poorly understood and mainly based on sporadic case reports/series. Our case presents a typical MLIII-gamma phenotype but with an unexpectedly early onset, expanding the clinical spectrum of this disease. It emphasizes the need for increased awareness among pediatric rheumatologists regarding metabolic disorders. Further case studies are essential to enhance understanding and improve diagnostic and therapeutic approaches for ML.
[Importance of lysosomal storage diseases in rheumatology].
Lysosomal storage diseases are a group of rare hereditary metabolic diseases. Due to a deficiency of lysosomal enzymes, complex substrates accumulate in the lysosomes of various organs. Depending on the affected enzyme, this results in clinically variable and chronic progressive multiorgan diseases. Diagnosis is often delayed. As clinical symptoms include the musculoskeletal system, an awareness of lysosomal storage diseases is of relevance to (pediatric) rheumatologists. This article is focused on Mucopolysaccharidosis type I‑S, Mucolipidosis type III, Gaucher disease and Fabry disease. When suspecting a lysosomal storage disease, enzyme activity should be determined in dried blood spots or leukocytes. For some diseases, specific biomarkers can additionally be analyzed. Diagnosis should be confirmed by genetic testing. As causal treatment options are available for three of the presented diseases, a timely diagnosis is very important. Lysosomale Speicherkrankheiten sind eine Gruppe seltener hereditärer Stoffwechselkrankheiten. Durch einen Mangel lysosomaler Enzyme akkumulieren komplexe Substrate in den Lysosomen verschiedener Organe. Abhängig vom betroffenen Enzym resultiert dies in klinisch variablen und chronisch progredienten Multiorgankrankheiten. Die Diagnosestellung erfolgt oft mit großer zeitlicher Verzögerung. Die klinische Symptomatik kann auch den Bewegungsapparat betreffen, weshalb eine grundlegende Kenntnis über lysosomale Speicherkrankheiten in der (kinder)rheumatologischen Praxis von Relevanz ist. Exemplarisch werden hier die Mukopolysaccharidose Typ I‑S, die Mukolipidose Typ III, der Morbus Gaucher und der Morbus Fabry vorgestellt. Bei Verdacht auf eine lysosomale Speicherkrankheit ist eine Bestimmung der Enzymaktivität im Trockenblut oder in Leukozyten angezeigt, teilweise lassen sich auch krankheitsspezifische Biomarker bestimmen. Zur Diagnosesicherung wird eine molekulargenetische Untersuchung ergänzt. Für 3 der hier vorgestellten Erkrankungen stehen kausale Therapien zur Verfügung, sodass der frühzeitigen Diagnosestellung eine große Bedeutung zukommt.
Unraveling mucolipidosis type III gamma through whole genome sequencing in late-onset retinitis pigmentosa: a case report.
We describe the case of a 47-year-old man referred to a retinal clinic and diagnosed with late-onset retinitis pigmentosa. Surprisingly, genetic testing revealed compound heterozygous pathogenic variants in GNPTG, leading to the diagnosis of the autosomal recessive lysosomal storage disorder mucolipidosis type III gamma. Mucolipidosis type III gamma is typically diagnosed during childhood due to symptoms relating to skeletal dysplasia. Retinal dystrophy is not a common phenotypic feature. Ophthalmologic examination was consistent with a mild form of retinitis pigmentosa and included fundus photography, measurement of best-corrected visual acuity, optical coherence tomography, electroretinogram and visual field testing. Extraocular findings included joint restriction and pains from an early age leading to bilateral hip replacement by age 30, aortic insufficiency, and hypertension. Genetic analysis was performed by whole genome sequencing filtered for a gene panel of 325 genes associated with retinal disease. Two compound heterozygous pathogenic variants were identified in GNPTG, c.347_349del and c.607dup. The diagnosis of mucolipidosis type III gamma was confirmed biochemically by measurement of increased activities of specific lysosomal enzymes in plasma. To our knowledge this is the first description of retinitis pigmentosa caused by compound heterozygous variants in GNPTG, providing further indications that late-onset retinal dystrophy is part of the phenotypic spectrum of mucolipidosis type III gamma.
Publicações recentes
When it's not juvenile idiopathic arthritis: unmasking monogenic mimickers in children monogenic mimickers of chronic arthritis.
Hand stiffness not only a rheumatological sign: A case of early onset mucolipidosis III-gamma with literature review.
Atypical presentation of mucolipidosis type III.
[Importance of lysosomal storage diseases in rheumatology].
Unraveling mucolipidosis type III gamma through whole genome sequencing in late-onset retinitis pigmentosa: a case report.
📚 EuropePMC32 artigos no totalmostrando 18
When it's not juvenile idiopathic arthritis: unmasking monogenic mimickers in children monogenic mimickers of chronic arthritis.
European journal of pediatricsHand stiffness not only a rheumatological sign: A case of early onset mucolipidosis III-gamma with literature review.
Molecular genetics and metabolism reportsAtypical presentation of mucolipidosis type III.
BMJ case reports[Importance of lysosomal storage diseases in rheumatology].
Zeitschrift fur RheumatologieUnraveling mucolipidosis type III gamma through whole genome sequencing in late-onset retinitis pigmentosa: a case report.
BMC ophthalmologyMucolipidosis: A mimicker of juvenile idiopathic arthritis.
International journal of rheumatic diseasesOrofacial abnormalities in mucopolysaccharidosis and mucolipidosis type II and III: A systematic review.
JIMD reportsUDP-GlcNAc-1-Phosphotransferase Is a Clinically Important Regulator of Human and Mouse Hair Pigmentation.
The Journal of investigative dermatologyImbalanced cellular metabolism compromises cartilage homeostasis and joint function in a mouse model of mucolipidosis type III gamma.
Disease models & mechanismsA Rare Manifestation of Right Ventricular Dysfunction in an Adult Patient With Mucolipidosis Type III α/β.
The Canadian journal of cardiologyDifferential diagnosis portfolio of a pediatric rheumatologist: eight cases, eight stories.
Clinical rheumatologyNewborn screening for Morquio disease and other lysosomal storage diseases: results from the 8-plex assay for 70,000 newborns.
Orphanet journal of rare diseasesMucolipidosis Type III: A Rare Disease in Differential Diagnosis of Joint Stiffness in Pediatric Rheumatology.
Archives of rheumatologyMucolipidosis type III, a series of adult patients.
Journal of inherited metabolic diseaseMucolipidosis type III gamma: Three novel mutation and genotype-phenotype study in eleven patients.
GeneNext Generation Sequencing identifies mutations in GNPTG gene as a cause of familial form of scleroderma-like disease.
Pediatric rheumatology online journalSolving a case of allelic dropout in the GNPTAB gene: implications in the molecular diagnosis of mucolipidosis type III alpha/beta.
Journal of pediatric endocrinology & metabolism : JPEMA novel splice site mutation in the GNPTAB gene in an Iranian patient with mucolipidosis II α/β.
Journal of pediatric endocrinology & metabolism : JPEMAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Mucolipidose tipo III.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Mucolipidose tipo III
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- When it's not juvenile idiopathic arthritis: unmasking monogenic mimickers in children monogenic mimickers of chronic arthritis.
- Atypical presentation of mucolipidosis type III.
- Hand stiffness not only a rheumatological sign: A case of early onset mucolipidosis III-gamma with literature review.
- [Importance of lysosomal storage diseases in rheumatology].
- Unraveling mucolipidosis type III gamma through whole genome sequencing in late-onset retinitis pigmentosa: a case report.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:577(Orphanet)
- OMIM OMIM:252600(OMIM)
- MONDO:0018931(MONDO)
- GARD:17704(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q56014231(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Mucolipidose tipo III
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata