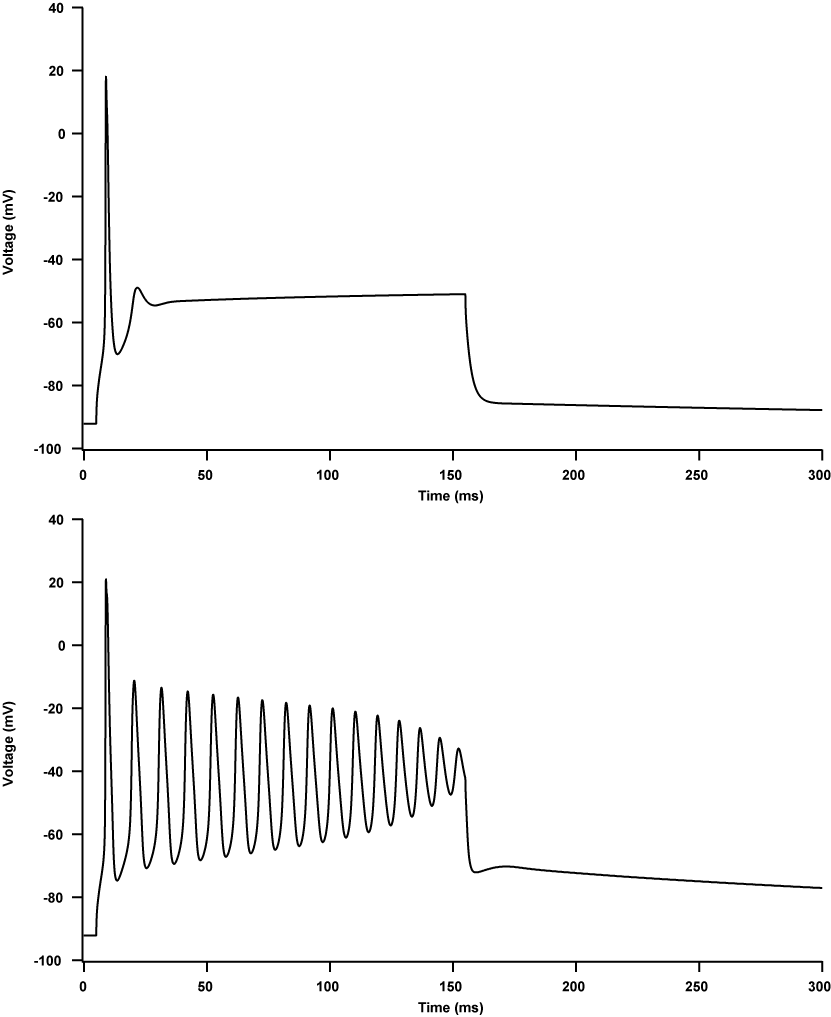
A miotonia agravada por potássio (PAM) é uma canalopatia muscular que se apresenta com miotonia pura dramaticamente agravada pela ingestão de potássio, com sensibilidade variável ao frio e sem fraqueza episódica. Este grupo inclui três formas: miotonia flutuante, miotonia permanente e miotonia responsiva à acetazolamida.
Introdução
O que você precisa saber de cara
A miotonia agravada por potássio (PAM) é uma canalopatia muscular que se apresenta com miotonia pura dramaticamente agravada pela ingestão de potássio, com sensibilidade variável ao frio e sem fraqueza episódica. Este grupo inclui três formas: miotonia flutuante, miotonia permanente e miotonia responsiva à acetazolamida.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 17 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 49 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisPore-forming subunit of Nav1.4, a voltage-gated sodium (Nav) channel that directly mediates the depolarizing phase of action potentials in excitable membranes. Navs, also called VGSCs (voltage-gated sodium channels) or VDSCs (voltage-dependent sodium channels), operate by switching between closed and open conformations depending on the voltage difference across the membrane. In the open conformation they allow Na(+) ions to selectively pass through the pore, along their electrochemical gradient.
Cell membrane
Paramyotonia congenita
An autosomal dominant channelopathy characterized by myotonia, increased by exposure to cold, intermittent flaccid paresis, not necessarily dependent on cold or myotonia, lability of serum potassium, non-progressive nature and lack of atrophy or hypertrophy of muscles. In some patients, myotonia is not increased by cold exposure (paramyotonia without cold paralysis). Patients may have a combination phenotype of PMC and HYPP.
Variantes genéticas (ClinVar)
488 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 605 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Miotonia agravada pelo potássio
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
Pesquisa e ensaios clínicos
2 ensaios clínicos encontrados.
Publicações mais relevantes
Piper rhythm-like electromyographical activity in muscle stiffness in sodium channel myotonia representing potassium-aggravated myotonia and myotonia permanens.
A 39-yr-old female with potassium-aggravated myotonia due to p.Q1633E in SCN4A experienced painful muscle stiffness triggered by exercise, potassium-rich fruits, and cold exposure, which progressed into a rigid state. Needle electromyography (EMG) during muscle stiffness showed synchronous, rhythmic, and regular activities starting at ∼60 Hz and ∼6 mV. A 37-yr-old male with myotonia permanens due to a splicing-affecting indel variant in intron 21 of SCN4A experienced cold-induced myotonia. EMG recordings during muscle stiffness showed similar synchronous, rhythmic, and regular activities starting at ∼80 Hz and 6.5 mV. In both patients, the frequencies and amplitudes were gradually decreased with relief of muscle stiffness. In either patient, single motor unit potentials by spontaneous activity were not explicitly recognized. In both patients, the activities produced a characteristic sound which, while similar in pitch to the "dive bomber" sound of classical myotonia, lacked its typical waxing and waning quality. The activities were similar to the Piper rhythm that was originally reported in fatigued normal muscle. Visual inspection of EMG activities in the literature revealed that similar Piper rhythm-like EMG activities were presented in Satoyoshi disease, myotonia permanens, paramyotonia congenita, and a rare form of nondystrophic myotonia. In Satoyoshi disease and fatigued normal skeletal muscle, the activities during muscle stiffness were less than 60 Hz, whereas in sodium channelopathies, they started at 60 Hz or higher, which may be a hallmark of hyperexcitability of the muscle membrane in sodium channelopathies.NEW & NOTEWORTHY In two patients with sodium channel myotonia representing potassium-aggravated myotonia and myotonia permanens, needle EMG showed Piper rhythm-like activities during muscle stiffness. Inspection of EMG recordings in the literature revealed similar Piper rhythm-like EMG activities in Satoyoshi disease, myotonia permanens, paramyotonia congenita, and a rare form of nondystrophic myotonia. Piper rhythm-like EMG activities starting at 60 Hz or higher, synchronizing with muscle stiffness, may be a hallmark of hyperexcitability of muscle membrane in sodium channelopathies.
Genetic analysis of a family with skeletal muscle ion channelopathy and hereditary spastic paraplegia type 7 caused by SCN4A and SPG7 double mutations.
Hereditary spastic paraplegia (HSP) is a rare and genetically heterogeneous neurodegenerative disorder, primarily defined by progressive lower-limb spasticity and weakness. Among the numerous genes implicated, pathogenic variants in the spastic paraplegia 7 (SPG7) gene represent one of the most common causes of HSP, whereas mutations in SCN4A, a skeletal muscle ion channel gene, are typically associated with a diverse spectrum of phenotypes, including hyperkalemic and hypokalemic periodic paralysis, potassium-aggravated myotonia, and congenital paramyotonia. To date, however, the coexistence of pathogenic variants in SPG7 and SCN4A within the same pedigree, and their potential pathogenic interplay, has not been documented. In this study, we performed comprehensive genetic profiling, including whole-exome sequencing, mitochondrial genome analysis, dynamic mutation screening, copy number variation assessment, and Sanger sequencing. We identified a novel heterozygous SPG7 variant (c.578A>G; p.E193G) alongside a known pathogenic SCN4A missense mutation (c.2111C>T; p.T704M). Remarkably, individuals harboring both variants presented with highly complex phenotypes that combined classical HSP manifestations with ion channel dysfunctions, such as congenital paramyotonia and hypokalemic periodic paralysis. These findings provide the first evidence of a possible genetic interaction between SPG7 and SCN4A, expanding the recognized clinical and molecular spectrum of HSP. Our results underscore the diagnostic value of multi-gene testing in patients with atypical or overlapping neuromuscular symptoms and highlight the importance of considering potential polygenic contributions when interpreting the clinical heterogeneity of HSP.
[Two cases of potassium-aggravated myotonia induced by SCN4A gene variation].
2例患儿为同胞姐弟,分别为7岁和2岁,均表现为发作性肢体活动障碍,感染、运动、食用香蕉后加重,幼时有发作性憋气、睁眼困难。基因检测发现2例患儿均为SCN4A基因22号外显子c.3917G>A(p.G1306E)发生错义变异,经基因深度测序发现患儿母亲该位点存在嵌合变异,变异率1.0236%,父亲为野生型,2例患儿确诊为钾加重性肌强直。给予低钾饮食、乙酰唑胺治疗,随诊8个月症状明显改善。.
Prevalence of genetically confirmed skeletal muscle channelopathies in the era of next generation sequencing.
We provide an up-to-date and accurate minimum point prevalence of genetically defined skeletal muscle channelopathies which is important for understanding the population impact, planning for treatment needs and future clinical trials. Skeletal muscle channelopathies include myotonia congenita (MC), sodium channel myotonia (SCM), paramyotonia congenita (PMC), hyperkalemic periodic paralysis (hyperPP), hypokalemic periodic paralysis (hypoPP) and Andersen- Tawil Syndrome (ATS). Patients referred to the UK national referral centre for skeletal muscle channelopathies and living in UK were included to calculate the minimum point prevalence using the latest data from the Office for National Statistics population estimate. We calculated a minimum point prevalence of all skeletal muscle channelopathies of 1.99/100 000 (95% CI 1.981-1.999). The minimum point prevalence of MC due to CLCN1 variants is 1.13/100 000 (95% CI 1.123-1.137), SCN4A variants which encode for PMC and SCM is 0.35/100 000 (95% CI 0.346 - 0.354) and for periodic paralysis (HyperPP and HypoPP) 0.41/100 000 (95% CI 0.406-0.414). The minimum point prevalence for ATS is 0.1/100 000 (95% CI 0.098-0.102). There has been an overall increase in point prevalence in skeletal muscle channelopathies compared to previous reports, with the biggest increase found to be in MC. This can be attributed to next generation sequencing and advances in clinical, electrophysiological and genetic characterisation of skeletal muscle channelopathies.
Clinical comparison and functional study of the L703P: a recurrent mutation in human SCN4A that causes sodium channel myotonia.
The non-dystrophic myotonias are inherited skeletal muscle disorders characterized by skeletal muscle stiffness after voluntary contraction, without muscle atrophy. Based on their clinical features, non-dystrophic myotonias are classified into myotonia congenita, paramyotonia congenita, and sodium channel myotonia. Using whole-exome next-generation sequencing, we identified a L703P mutation (c.2108T>C, p.L703P) in SCN4A in a Chinese family diagnosed with non-dystrophic myotonias. The clinical findings of patients in this family included muscle stiffness and hypertrophy. The biophysical properties of wildtype and mutant channels were investigated using whole-cell patch clamp. L703P causes both gain-of-function and loss-of-function changes in Nav1.4 properties, including decreased current density, impaired recovery, enhanced activation and slow inactivation. Our study demonstrates that L703P is a pathogenic variant for myotonia, and provides additional electrophysiological information for understanding the pathogenic mechanism of SCN4A-associated channelopathies.
Publicações recentes
Piper rhythm-like electromyographical activity in muscle stiffness in sodium channel myotonia representing potassium-aggravated myotonia and myotonia permanens.
Genetic analysis of a family with skeletal muscle ion channelopathy and hereditary spastic paraplegia type 7 caused by SCN4A and SPG7 double mutations.
[Two cases of potassium-aggravated myotonia induced by SCN4A gene variation].
"Status myotonicus" in Na(v)1.4-M1592V channelopathy.
Anesthetic management of a patient with sodium-channel myotonia: a case report.
📚 EuropePMC4 artigos no totalmostrando 20
Piper rhythm-like electromyographical activity in muscle stiffness in sodium channel myotonia representing potassium-aggravated myotonia and myotonia permanens.
Journal of neurophysiologyGenetic analysis of a family with skeletal muscle ion channelopathy and hereditary spastic paraplegia type 7 caused by SCN4A and SPG7 double mutations.
Gene[Two cases of potassium-aggravated myotonia induced by SCN4A gene variation].
Zhonghua er ke za zhi = Chinese journal of pediatricsPrevalence of genetically confirmed skeletal muscle channelopathies in the era of next generation sequencing.
Neuromuscular disorders : NMDClinical comparison and functional study of the L703P: a recurrent mutation in human SCN4A that causes sodium channel myotonia.
Neuromuscular disorders : NMD"Status myotonicus" in Nav1.4-M1592V channelopathy.
Neuromuscular disorders : NMDGuidelines on clinical presentation and management of nondystrophic myotonias.
Muscle & nerveAnesthetic management of a patient with sodium-channel myotonia: a case report.
JA clinical reportsDe novo variant in SCN4A causes neonatal sodium channel myotonia with general muscle stiffness and respiratory failure.
Neuromuscular disorders : NMDA zebrafish model of nondystrophic myotonia with sodium channelopathy.
Neuroscience lettersSAR inspired by aldehyde oxidase (AO) metabolism: Discovery of novel, CNS penetrant tricyclic M4 PAMs.
Bioorganic & medicinal chemistry lettersHerculean Boy With Facial Myokymia.
Pediatric neurologyMyotonia permanens with Nav1.4-G1306E displays varied phenotypes during course of life.
Acta myologica : myopathies and cardiomyopathies : official journal of the Mediterranean Society of MyologyIn vivo assessment of muscle membrane properties in the sodium channel myotonias.
Muscle & nerve[An unusual case of sodium channel myotonia with transient weakness upon initiating movements which is characteristic in Becker disease].
Rinsho shinkeigaku = Clinical neurologyA Sodium Channel Myotonia Presenting with Intermittent Dysphagia as a Manifestation of a Rare SCN4A Variant.
Journal of molecular neuroscience : MNTranslational approach to address therapy in myotonia permanens due to a new SCN4A mutation.
NeurologyFlecainide-Responsive Myotonia Permanens With SNEL Onset: A New Case and Literature Review.
PediatricsPainful cramps and giant myotonic discharges in a family with the Nav1.4-G1306A mutation.
Muscle & nerveNew phenotype and neonatal onset of sodium channel myotonia in a child with a novel mutation of SCN4A gene.
Brain & developmentAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Miotonia agravada pelo potássio.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Miotonia agravada pelo potássio
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Piper rhythm-like electromyographical activity in muscle stiffness in sodium channel myotonia representing potassium-aggravated myotonia and myotonia permanens.
- Genetic analysis of a family with skeletal muscle ion channelopathy and hereditary spastic paraplegia type 7 caused by SCN4A and SPG7 double mutations.
- [Two cases of potassium-aggravated myotonia induced by SCN4A gene variation].
- Prevalence of genetically confirmed skeletal muscle channelopathies in the era of next generation sequencing.
- Clinical comparison and functional study of the L703P: a recurrent mutation in human SCN4A that causes sodium channel myotonia.
- "Status myotonicus" in Na(v)1.4-M1592V channelopathy.
- Anesthetic management of a patient with sodium-channel myotonia: a case report.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:612(Orphanet)
- OMIM OMIM:608390(OMIM)
- MONDO:0018959(MONDO)
- GARD:4459(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q7234683(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Miotonia agravada pelo potássio
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata