A Disostose Acrofacial, tipo Weyers (WAD), é uma síndrome rara que afeta o desenvolvimento de tecidos como a pele, cabelo, unhas e dentes, além de causar problemas nos ossos. Ela é caracterizada por unhas com deformidades; anomalias no queixo, na parte da boca entre os dentes e os lábios/bochechas (vestíbulo oral), e nos dentes; a presença de dedos extras nas mãos ou pés (polidactilia pós-axial), geralmente no lado do dedo mínimo; crescimento moderadamente limitado com braços e pernas mais curtos; e inteligência normal. Embora se assemelhe muito à síndrome de Ellis-van Creveld — que é causada por um problema no mesmo gene e também é um tipo de "ciliopatia" (doença que afeta pequenas estruturas celulares chamadas cílios) —, a WAD costuma ser uma condição mais leve, que não apresenta problemas no coração. Além disso, ela é transmitida geneticamente por herança autossômica dominante, o que significa que basta herdar uma cópia alterada do gene de um dos pais para desenvolver a doença.
Introdução
O que você precisa saber de cara
A Disostose Acrofacial, tipo Weyers (WAD), é uma síndrome rara que afeta o desenvolvimento de tecidos como a pele, cabelo, unhas e dentes, além de causar problemas nos ossos. Ela é caracterizada por unhas com deformidades; anomalias no queixo, na parte da boca entre os dentes e os lábios/bochechas (vestíbulo oral), e nos dentes; a presença de dedos extras nas mãos ou pés (polidactilia pós-axial), geralmente no lado do dedo mínimo; crescimento moderadamente limitado com braços e pernas mais curtos; e inteligência normal. Embora se assemelhe muito à síndrome de Ellis-van Creveld — que é causada por um problema no mesmo gene e também é um tipo de "ciliopatia" (doença que afeta pequenas estruturas celulares chamadas cílios) —, a WAD costuma ser uma condição mais leve, que não apresenta problemas no coração. Além disso, ela é transmitida geneticamente por herança autossômica dominante, o que significa que basta herdar uma cópia alterada do gene de um dos pais para desenvolver a doença.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 9 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 26 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
3 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisComponent of the EvC complex that positively regulates ciliary Hedgehog (Hh) signaling. Plays a critical role in bone formation and skeletal development. May be involved in early embryonic morphogenesis
Cell membraneCytoplasm, cytoskeleton, cilium basal bodyCell projection, ciliumCell projection, cilium membraneNucleus
Ellis-van Creveld syndrome
An autosomal recessive condition characterized by the clinical tetrad of chondrodystrophy, polydactyly, ectodermal dysplasia and cardiac anomalies. Patients manifest short-limb dwarfism, short ribs, postaxial polydactyly, and dysplastic nails and teeth. Congenital heart defects, most commonly an atrioventricular septal defect, are observed in 60% of affected individuals.
Component of the EvC complex that positively regulates ciliary Hedgehog (Hh) signaling. Involved in endochondral growth and skeletal development
Cell membraneCytoplasm, cytoskeleton, cilium basal bodyCell projection, ciliumCell projection, cilium membrane
Ellis-van Creveld syndrome
An autosomal recessive condition characterized by the clinical tetrad of chondrodystrophy, polydactyly, ectodermal dysplasia and cardiac anomalies. Patients manifest short-limb dwarfism, short ribs, postaxial polydactyly, and dysplastic nails and teeth. Congenital heart defects, most commonly an atrioventricular septal defect, are observed in 60% of affected individuals.
Key downstream component of the canonical Wnt signaling pathway (PubMed:17524503, PubMed:18077326, PubMed:18086858, PubMed:18957423, PubMed:21262353, PubMed:22155184, PubMed:22647378, PubMed:22699938). In the absence of Wnt, forms a complex with AXIN1, AXIN2, APC, CSNK1A1 and GSK3B that promotes phosphorylation on N-terminal Ser and Thr residues and ubiquitination of CTNNB1 via BTRC and its subsequent degradation by the proteasome (PubMed:17524503, PubMed:18077326, PubMed:18086858, PubMed:189574
CytoplasmNucleusCytoplasm, cytoskeletonCell junction, adherens junctionCell junctionCell membraneCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, spindle poleSynapseCytoplasm, cytoskeleton, cilium basal body
Colorectal cancer
A complex disease characterized by malignant lesions arising from the inner wall of the large intestine (the colon) and the rectum. Genetic alterations are often associated with progression from premalignant lesion (adenoma) to invasive adenocarcinoma. Risk factors for cancer of the colon and rectum include colon polyps, long-standing ulcerative colitis, and genetic family history.
Variantes genéticas (ClinVar)
1.331 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
32 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Disostose acrofacial, tipo Weyers
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
A novel EVC2 splice-site variant expands the mutational and phenotypic spectrum of Weyers acrofacial dysostosis.
Variant characterisation and clinical profile in a large cohort of patients with Ellis-van Creveld syndrome and a family with Weyers acrofacial dysostosis.
Ellis-van Creveld syndrome (EvC) is a recessive disorder characterised by acromesomelic limb shortening, postaxial polydactyly, nail-teeth dysplasia and congenital cardiac defects, primarily caused by pathogenic variants in EVC or EVC2. Weyers acrofacial dysostosis (WAD) is an ultra-rare dominant condition allelic to EvC. The present work aimed to enhance current knowledge on the clinical manifestations of EvC and WAD and broaden their mutational spectrum. We conducted molecular studies in 46 individuals from 43 unrelated families with a preliminary clinical diagnosis of EvC and 3 affected individuals from a family with WAD and retrospectively analysed clinical data. The deleterious effect of selected variants of uncertain significance was evaluated by cellular assays. We identified pathogenic variants in EVC/EVC2 in affected individuals from 41 of the 43 families with EvC. Patients from each of the two remaining families were found with a homozygous splicing variant in WDR35 and a de novo heterozygous frameshift variant in GLI3, respectively. The phenotype of these patients showed a remarkable overlap with EvC. A novel EVC2 C-terminal truncating variant was identified in the family with WAD. Deep phenotyping of the cohort recapitulated 'classical EvC findings' in the literature and highlighted findings previously undescribed or rarely described as part of EvC. This study presents the largest cohort of living patients with EvC to date, contributing to better understanding of the full clinical spectrum of EvC. We also provide comprehensive information on the EVC/EVC2 mutational landscape and add GLI3 to the list of genes associated with EvC-like phenotypes.
Weyers Acrofacial Dysostosis: A Case Report.
Weyers acrofacial dysostosis (WAD) is a rare skeletal dysplasia, which is autosomal-dominant, and the clinical symptoms are presented as dental anomalies, polydactyly, nail dystrophy, and short physical stature. It is also termed "Curry‑Hall syndrome" and reported to be linked to genetic mutations mapped on chromosome 4p16, the region reported being commonly associated with a similar genetic syndrome, Ellis-van Creveld (EVC) syndrome. Most individuals with EVC have congenital heart abnormalities, most often atrial septal defects, unlike WAD. In this case, a 15‑year‑old girl presented with onychodystrophy and polydactyly observed in the hands and feet, microdontia, or agenesis of teeth, which were conical in shape, with a short stature. The patient had dystrophy of nails since birth, and physical growth in terms of height did not match the normal growth parameters with respect to age. The patient also had abnormal dentation with conical-shaped teeth, with the rest of the clinical presentations suggestive of WAD.
EVC-EVC2 complex stability and ciliary targeting are regulated by modification with ubiquitin and SUMO.
Ellis van Creveld syndrome and Weyers acrofacial dysostosis are two rare genetic diseases affecting skeletal development. They are both ciliopathies, as they are due to malfunction of primary cilia, microtubule-based plasma membrane protrusions that function as cellular antennae and are required for Hedgehog signaling, a key pathway during skeletal morphogenesis. These ciliopathies are caused by mutations affecting the EVC-EVC2 complex, a transmembrane protein heterodimer that regulates Hedgehog signaling from inside primary cilia. Despite the importance of this complex, the mechanisms underlying its stability, targeting and function are poorly understood. To address this, we characterized the endogenous EVC protein interactome in control and Evc-null cells. This proteomic screen confirmed EVC's main known interactors (EVC2, IQCE, EFCAB7), while revealing new ones, including USP7, a deubiquitinating enzyme involved in Hedgehog signaling. We therefore looked at EVC-EVC2 complex ubiquitination. Such ubiquitination exists but is independent of USP7 (and of USP48, also involved in Hh signaling). We did find, however, that monoubiquitination of EVC-EVC2 cytosolic tails greatly reduces their protein levels. On the other hand, modification of EVC-EVC2 cytosolic tails with the small ubiquitin-related modifier SUMO3 has a different effect, enhancing complex accumulation at the EvC zone, immediately distal to the ciliary transition zone, possibly via increased binding to the EFCAB7-IQCE complex. Lastly, we find that EvC zone targeting of EVC-EVC2 depends on two separate EFCAB7-binding motifs within EVC2's Weyers-deleted peptide. Only one of these motifs had been characterized previously, so we have mapped the second herein. Altogether, our data shed light on EVC-EVC2 complex regulatory mechanisms, with implications for ciliopathies.
Microdeletion of 4p16.2 in Children: A Case Report and Literature Review.
Copy number variations (CNV) are thought to play an important role in causing human diseases, including congenital anomalies, psychiatric disorders, and intellectual disabilities. We report here a one-year-old boy presented to our clinic as developmental delay. He presented a birth weight of 4.5 kg, motor delay, mental retardation, mild hypertonia, and some dysmorphic features (mild frontal bossing, hypertelorism, epicanthus, concave nasal ridge, slightly sparse hair, short hands, and mild nail dysplasia). The brain MRI indicated brain abnormalities; the Gross Motor Function Measure-66 score was 23.37; the Gesell test result showed the development quotient was 50, suggesting mental retardation. Chromosomal microarray analysis showed an approximately 97 kb microdeletion at 4p16.2 (4p16.2 CNV), including part of EVC and EVC2 genes, which were associated with Ellis-van Creveld syndrome (EvC) and Weyers acrofacial dysostosis (WAD). This report suggests 4p16.2 microdeletion may be associated with multiple developmental abnormalities, including motor delay and mental retardation.
Publicações recentes
A novel EVC2 splice-site variant expands the mutational and phenotypic spectrum of Weyers acrofacial dysostosis.
A New Case of Nager Syndrome as a Rare Cause of Acrofacial Dysostosis.
The Phenotypic Spectrum of Miller Syndrome: Insight From a French Cohort.
RNA Polymerase I Dysfunction Underlying Craniofacial Syndromes: Integrated Genetic Analysis Reveals Parallels to 22q11.2 Deletion Syndrome.
Facial Bone Defects Associated with Lateral Facial Clefts Tessier Type 6, 7 and 8 in Syndromic Neurocristopathies: A Detailed Micro-CT Analysis on Historical Museum Specimens.
📚 EuropePMCmostrando 9
A novel EVC2 splice-site variant expands the mutational and phenotypic spectrum of Weyers acrofacial dysostosis.
BMC medical genomicsVariant characterisation and clinical profile in a large cohort of patients with Ellis-van Creveld syndrome and a family with Weyers acrofacial dysostosis.
Journal of medical geneticsWeyers Acrofacial Dysostosis: A Case Report.
CureusEVC-EVC2 complex stability and ciliary targeting are regulated by modification with ubiquitin and SUMO.
Frontiers in cell and developmental biologyMicrodeletion of 4p16.2 in Children: A Case Report and Literature Review.
Case reports in geneticsNovel mutation in EFCAB7 alters expression and interaction of Ellis-van Creveld ciliary proteins.
Congenital anomaliesRole of Primary Cilia in Odontogenesis.
Journal of dental research[Analysis of causes and whole microbial structure in a case of rampant caries].
Nan fang yi ke da xue xue bao = Journal of Southern Medical UniversityNovel mutations in EVC cause aberrant splicing in Ellis-van Creveld syndrome.
Molecular genetics and genomics : MGGAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Disostose acrofacial, tipo Weyers.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Disostose acrofacial, tipo Weyers
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- A novel EVC2 splice-site variant expands the mutational and phenotypic spectrum of Weyers acrofacial dysostosis.
- Variant characterisation and clinical profile in a large cohort of patients with Ellis-van Creveld syndrome and a family with Weyers acrofacial dysostosis.
- Weyers Acrofacial Dysostosis: A Case Report.
- EVC-EVC2 complex stability and ciliary targeting are regulated by modification with ubiquitin and SUMO.
- Microdeletion of 4p16.2 in Children: A Case Report and Literature Review.
- A New Case of Nager Syndrome as a Rare Cause of Acrofacial Dysostosis.
- The Phenotypic Spectrum of Miller Syndrome: Insight From a French Cohort.
- RNA Polymerase I Dysfunction Underlying Craniofacial Syndromes: Integrated Genetic Analysis Reveals Parallels to 22q11.2 Deletion Syndrome.
- Facial Bone Defects Associated with Lateral Facial Clefts Tessier Type 6, 7 and 8 in Syndromic Neurocristopathies: A Detailed Micro-CT Analysis on Historical Museum Specimens.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:952(Orphanet)
- OMIM OMIM:193530(OMIM)
- MONDO:0008673(MONDO)
- GARD:497(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55781611(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
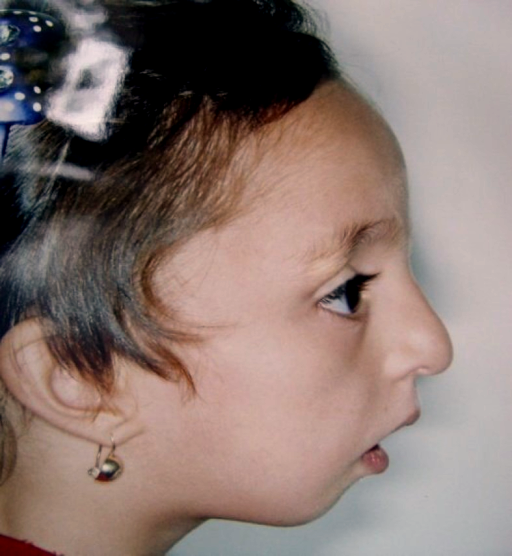
Disostose acrofacial, tipo Weyers
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata