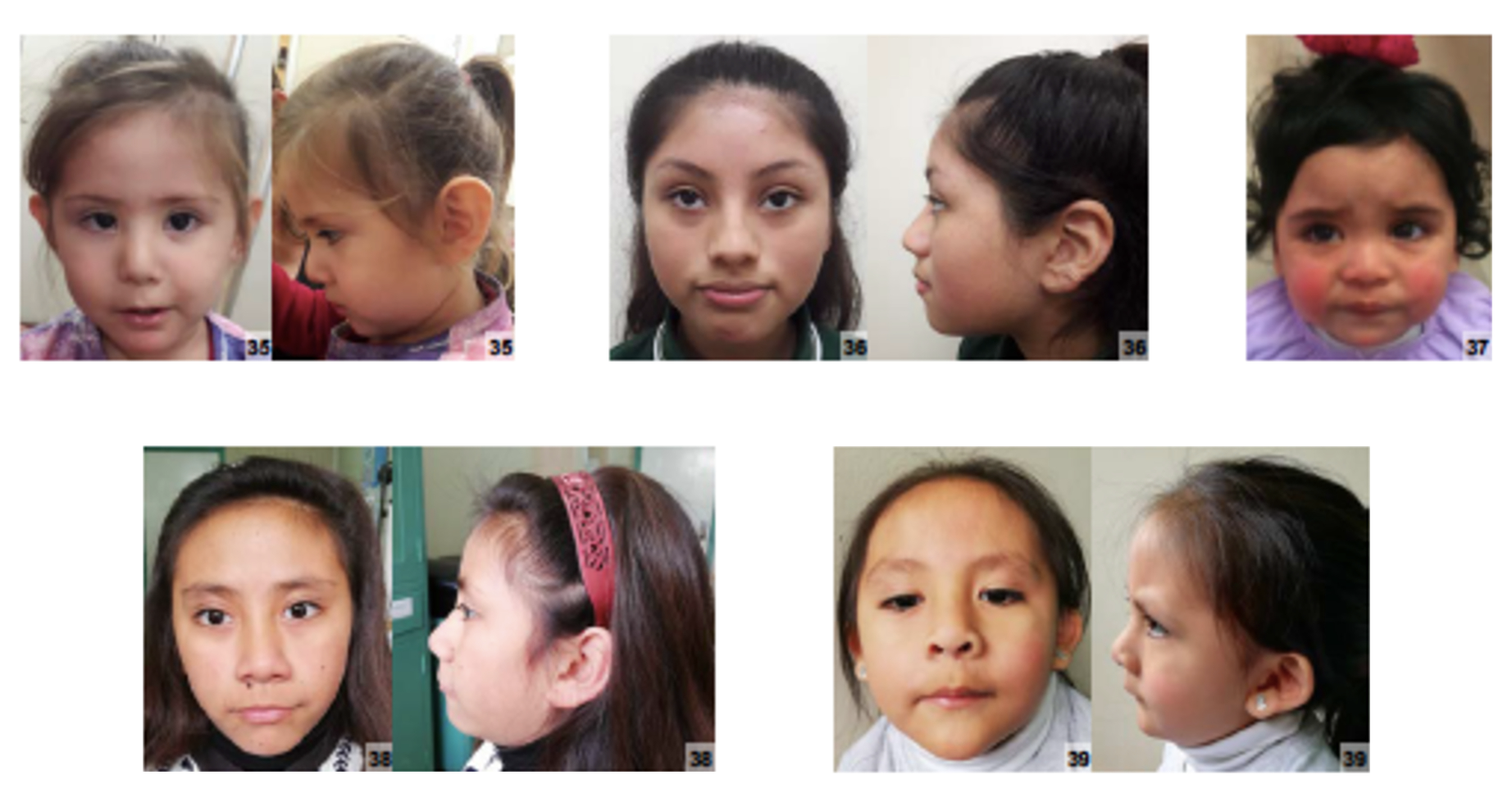
Esta síndrome se manifesta pela combinação de: ausência completa de cabelos (presente desde o nascimento), deficiência intelectual leve e um problema nos ovários ou testículos que não funcionam adequadamente, mesmo com o corpo produzindo muitos hormônios para tentar estimular seu funcionamento.
Introdução
O que você precisa saber de cara
Esta síndrome se manifesta pela combinação de: ausência completa de cabelos (presente desde o nascimento), deficiência intelectual leve e um problema nos ovários ou testículos que não funcionam adequadamente, mesmo com o corpo produzindo muitos hormônios para tentar estimular seu funcionamento.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 3 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de alopecia-transtorno do desenvolvimento intelectual-hipogonadismo hipergonadotrópico
Centros de Referência SUS
13 centros habilitados pelo SUS para Síndrome de alopecia-transtorno do desenvolvimento intelectual-hipogonadismo hipergonadotrópico
Centros para Síndrome de alopecia-transtorno do desenvolvimento intelectual-hipogonadismo hipergonadotrópico
Detalhes dos centros
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
3 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Mostrando amostra de 2 publicações de um total de 7.533
Hypotrichosis 14: novel variants of the LSS gene in five Chinese families and insights from literature review.
Congenital hypotrichosis 14 is a nonsyndromic form of alopecia associated with pathogenic variants in the lanosterol synthase (LSS) gene. Recent studies have expanded the spectrum of LSS-related phenotypes, including congenital cataracts, alopecia-intellectual disability syndrome, and palmoplantar keratoderma. Currently, investigations into this disease are still limited, and its treatment remains elusive. In this study, we aimed to report six Chinese patients who were diagnosed with hypotrichosis 14, whose conditions were attributed to five novel and recurrent variants in the LSS gene identified via whole exome sequencing. Moreover, the reported LSS gene has also been summarized. We described six patients in five Chinese families with hair loss, and one of whom had a rare accompanying phenotype of hypergonadotropic hypogonadism, which has rarely been reported. Five novel variants were discovered in the LSS gene, including c.919_921del (p.His307del), c.1987 C > T (p.Arg663Trp), c.982 C > T (p.Arg328*), c.1405_1407del (p.Glu469del) and c.193_200dup (p.Pro68Argfs*14). Our comprehensive summary of phenotypes caused by LSS gene variants not only enriches the existing knowledge on congenital hypotrichosis 14 but also provides crucial guidance for more accurate genetic counseling and potentially new directions for future research in understanding the disease mechanism and developing targeted therapies.
The Use of High-Density SNP Array to Map Homozygosity in Consanguineous Families to Efficiently Identify Candidate Genes: Application to Woodhouse-Sakati Syndrome.
Two consanguineous Qatari siblings presented for evaluation: a 17-4/12-year-old male with hypogonadotropic hypogonadism, alopecia, intellectual disability, and microcephaly and his 19-year-old sister with primary amenorrhea, alopecia, and normal cognition. Both required hormone treatment to produce secondary sex characteristics and pubertal development beyond Tanner 1. SNP array analysis of both probands was performed to detect shared regions of homozygosity which may harbor homozygous mutations in a gene causing their common features of abnormal pubertal development, alopecia, and variable cognitive delay. Our patients shared multiple homozygous genomic regions; ten shared regions were >1 Mb in length and constituted 0.99% of the genome. DCAF17, encoding a transmembrane nuclear protein of uncertain function, was the only gene identified in a homozygous region known to cause hypogonadotropic hypogonadism. DCAF17 mutations are associated with Woodhouse-Sakati syndrome, a rare disorder characterized by alopecia, hypogonadotropic hypogonadism, sensorineural hearing loss, diabetes mellitus, and extrapyramidal movements. Sequencing of the coding exons and flanking intronic regions of DCAF17 in the proband revealed homozygosity for a previously described founder mutation (c.436delC). Targeted DCAF17 sequencing of his affected sibling revealed the same homozygous mutation. This family illustrates the utility of SNP array testing in consanguineous families to efficiently and inexpensively identify regions of genomic homozygosity in which genetic candidates for recessive conditions can be identified.
Publicações recentes
Abduction-Release Sign in Heavy Eye Syndrome.
Anti-GQ1b-Positive Miller Fisher Syndrome Following Pfizer Bivalent COVID-19 Vaccination.
A review of chronic enterocolitis of rhesus macaques (Macaca mulatta) and potential as a naturally occurring model for post-infectious irritable bowel syndrome.
Ophthalmic manifestations and management of Traboulsi syndrome in three children of a Saudi family.
Potential mechanisms of the glucocorticoid withdrawal syndrome.
📚 EuropePMCmostrando 2
Hypotrichosis 14: novel variants of the LSS gene in five Chinese families and insights from literature review.
Human genomicsThe Use of High-Density SNP Array to Map Homozygosity in Consanguineous Families to Efficiently Identify Candidate Genes: Application to Woodhouse-Sakati Syndrome.
Case reports in geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de alopecia-transtorno do desenvolvimento intelectual-hipogonadismo hipergonadotrópico.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de alopecia-transtorno do desenvolvimento intelectual-hipogonadismo hipergonadotrópico
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Hypotrichosis 14: novel variants of the LSS gene in five Chinese families and insights from literature review.
- The Use of High-Density SNP Array to Map Homozygosity in Consanguineous Families to Efficiently Identify Candidate Genes: Application to Woodhouse-Sakati Syndrome.
- Abduction-Release Sign in Heavy Eye Syndrome.
- Anti-GQ1b-Positive Miller Fisher Syndrome Following Pfizer Bivalent COVID-19 Vaccination.
- A review of chronic enterocolitis of rhesus macaques (Macaca mulatta) and potential as a naturally occurring model for post-infectious irritable bowel syndrome.
- Ophthalmic manifestations and management of Traboulsi syndrome in three children of a Saudi family.
- Potential mechanisms of the glucocorticoid withdrawal syndrome.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1014(Orphanet)
- OMIM OMIM:601217(OMIM)
- MONDO:0011019(MONDO)
- GARD:16553(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Q55783029(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome de alopecia-transtorno do desenvolvimento intelectual-hipogonadismo hipergonadotrópico
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata