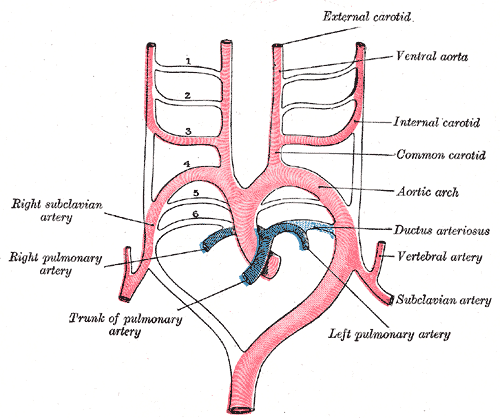
As malformações congênitas (presentes desde o nascimento) do arco da aorta (a principal artéria que sai do coração) acontecem quando uma ou mais partes do sistema de arcos faríngeos embrionários (estruturas que se formam na região da garganta do bebê ainda na barriga da mãe) não se desenvolvem corretamente. Qualquer parte desse sistema pode desaparecer ou permanecer de forma errada, o que gera uma grande variedade de problemas no arco da aorta. Do ponto de vista médico, essas malformações são divididas em dois tipos: aquelas que causam (ou têm grande chance de causar) problemas no funcionamento do corpo e aquelas que não causam. Esses problemas no funcionamento do corpo incluem a compressão da traqueia e dos brônquios (os "tubos" por onde o ar passa para os pulmões), a compressão do esôfago (o "tubo" por onde a comida passa para o estômago) e padrões incomuns no fluxo de sangue.
Introdução
O que você precisa saber de cara
As malformações congênitas (presentes desde o nascimento) do arco da aorta (a principal artéria que sai do coração) acontecem quando uma ou mais partes do sistema de arcos faríngeos embrionários (estruturas que se formam na região da garganta do bebê ainda na barriga da mãe) não se desenvolvem corretamente. Qualquer parte desse sistema pode desaparecer ou permanecer de forma errada, o que gera uma grande variedade de problemas no arco da aorta. Do ponto de vista médico, essas malformações são divididas em dois tipos: aquelas que causam (ou têm grande chance de causar) problemas no funcionamento do corpo e aquelas que não causam. Esses problemas no funcionamento do corpo incluem a compressão da traqueia e dos brônquios (os "tubos" por onde o ar passa para os pulmões), a compressão do esôfago (o "tubo" por onde a comida passa para o estômago) e padrões incomuns no fluxo de sangue.
Tem tratamento?
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 30 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 54 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
3 genes identificados com associação a esta condição. Padrão de herança: Not applicable.
Transcription regulator. Forms a sequence-specific DNA-binding protein complex with MAD1, MAD4, MNT, WBSCR14 and MLXIP which recognizes the core sequence 5'-CACGTG-3'. The TCFL4-MAD1, TCFL4-MAD4, TCFL4-WBSCR14 complexes are transcriptional repressors. Plays a role in transcriptional activation of glycolytic target genes. Involved in glucose-responsive gene regulation
CytoplasmNucleus
Cytokine that can act as a growth factor for activated T and NK cells, enhance the lytic activity of NK/lymphokine-activated killer cells, and stimulate the production of IFN-gamma by resting PBMC Associates with IL23A to form the IL-23 interleukin, a heterodimeric cytokine which functions in innate and adaptive immunity. IL-23 may constitute with IL-17 an acute response to infection in peripheral tissues. IL-23 binds to a heterodimeric receptor complex composed of IL12RB1 and IL23R, activates t
Secreted
Immunodeficiency 29
A form of Mendelian susceptibility to mycobacterial disease, a rare condition caused by impairment of interferon-gamma mediated immunity. It is characterized by predisposition to illness caused by moderately virulent mycobacterial species, such as Bacillus Calmette-Guerin (BCG) vaccine, environmental non-tuberculous mycobacteria, and by the more virulent Mycobacterium tuberculosis. Other microorganisms rarely cause severe clinical disease in individuals with susceptibility to mycobacterial infections, with the exception of Salmonella which infects less than 50% of these individuals. Clinical outcome severity depends on the degree of impairment of interferon-gamma mediated immunity. Some patients die of overwhelming mycobacterial disease with lepromatous-like lesions in early childhood, whereas others develop, later in life, disseminated but curable infections with tuberculoid granulomas. IMD29 is characterized by undetectable IL12B secretion from leukocytes. Affected individuals generally present with BCG disease after vaccination in childhood, and at least half also have Salmonella infection. Disease phenotype is relatively mild, and patients have a good prognosis.
Antigen-presenting major histocompatibility complex class I (MHCI) molecule. In complex with B2M/beta 2 microglobulin displays primarily viral and tumor-derived peptides on antigen-presenting cells for recognition by alpha-beta T cell receptor (TCR) on HLA-B-restricted CD8-positive T cells, guiding antigen-specific T cell immune response to eliminate infected or transformed cells (PubMed:23209413, PubMed:25808313, PubMed:29531227, PubMed:9620674). May also present self-peptides derived from the
Cell membraneEndoplasmic reticulum membrane
Stevens-Johnson syndrome
A rare blistering mucocutaneous disease that share clinical and histopathologic features with toxic epidermal necrolysis. Both disorders are characterized by high fever, malaise, and a rapidly developing blistering exanthema of macules and target-like lesions accompanied by mucosal involvement. Stevens-Johnson syndrome is a milder disease characterized by destruction and detachment of the skin epithelium and mucous membranes involving less than 10% of the body surface area. Ocular symptoms include ulcerative conjunctivitis, keratitis, iritis, uveitis and sometimes blindness. It can be caused by a severe adverse reaction to particular types of medication, although Mycoplasma infections may induce some cases.
Medicamentos e terapias
Mecanismo: Interleukin-6 receptor alpha subunit inhibitor
Mecanismo: Inosine-5'-monophosphate dehydrogenase (IMPDH) inhibitor
Mecanismo: Amidophosphoribosyltransferase inhibitor
Mecanismo: Janus Kinase (JAK) inhibitor
Mecanismo: Tyrosine-protein kinase JAK2 inhibitor
Mecanismo: Interleukin-23 inhibitor
Mecanismo: Glucocorticoid receptor agonist
Mecanismo: TNF-alpha inhibitor
Mecanismo: T-lymphocyte activation antigen CD86 inhibitor
Mecanismo: Glucocorticoid receptor agonist
Variantes genéticas (ClinVar)
62 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
14 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Anomalia do arco aórtico
Centros de Referência SUS
24 centros habilitados pelo SUS para Anomalia do arco aórtico
Centros para Anomalia do arco aórtico
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
3 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Comprehensive defect detection in mouse embryos and the heart by combining automated phenotyping with novel population averages and atlases.
Micro-CT has become the standard for the assessment of malformations in mouse embryos because it allows the visualisation of internal structures in the context of the intact embryo. Statistical comparison of volume differences is possible via manual segmentation of organs of interest from micro-CT scans, but this process is slow and laborious. Automated registration-based methods now exist that make the volumetric analysis of all organs feasible. Here, we expand the available atlases for use with the LAMA registration and analysis pipeline to include high-resolution population averages derived from phosphotungstic acid-stained C57BL/6J embryos and corresponding manually segmented atlases at embryonic stage (E) 12.5, E15.5, and E17.5. We report application of these population averages and atlases with the LAMA phenotyping pipeline to Wbp11 heterozygous null embryos, identifying defects previously reported in the cervical vertebrae, brain, nasal cavity, palate, liver and kidneys as well as a right aortic arch defects missed by manual analysis, and volume differences in the eyes and spinal cord. Finally, we report a high-resolution isolated E18.5 mouse heart population average and corresponding atlas that when applied to the Wbp11 line identified significant differences. These findings highlight the advantages of unbiased, volumetric and quantitative approaches in the analysis of mouse models of human disease.
Maternal Genotype and Dietary Vitamin A Modify Aortic Arch Phenotypes in a Mouse Model of 22q11DS.
Congenital heart defects (CHDs) occur in 50-75% of patients with 22q11.2 deletion syndrome (22q11.2DS), ranging from mild to severe manifestations. The genetic and environmental factors contributing to variable CHD phenotypes in 22q11.2DS are largely unknown. In this study, we used a mouse model of 22q11.2DS, termed Df1/+, to evaluate the effect of maternal vitamin A (VitA) dietary imbalance (supplementation or deficiency) on the incidence of aortic arch defects (AADs), which is a common type of CHD observed in both 22q11.2DS patients and Df1/+ mouse embryos. While most groups showed a previously observed 30% AAD incidence, two groups exhibited significantly higher rates: (1) Df1/+ embryos from WT mothers on a VitA-Supl diet (51% AADs) and (2) Df1/+ embryos from Df1/+ mothers on a VitA-Def diet (45% AADs). Thus, a low or high maternal VitA diet can increase the frequency of AADs in embryos depending on the maternal genotype. Transcriptomic analysis of the hearts of these high-risk embryos at embryonic day (E)18.5 revealed downregulation of key genes (Hdac3, Ptgds, Sirt5, Pfkm, and Lclat1) associated with energy metabolism pathways, such as oxidative phosphorylation and glycolysis, suggesting impaired cardiac recovery mechanisms. In conclusion, our findings demonstrate that altered VitA exposure can exacerbate AAD incidence in a maternal-genotype-dependent manner, highlighting the complex interplay between embryonic and maternal genetic background and environmental factors in CHDs associated with 22q11.2DS.
Hybrid Procedure for Coexistence of Coarctation of the Aorta and Aberrant Right Subclavian Artery: A Case Report and Review of the Literature.
Background: Coarctation of the aorta (CoA) can either present alone as an isolated condition or in association with other aortic arch or cardiac anomalies. One percent of patients with CoA have concomitant an aberrant right subclavian artery (ARSA). Purpose: We report the case of a 35-year-old woman with uncontrolled hypertension who was found to have CoA and ARSA. Results: The patient was treated successfully using a hybrid procedure comprising ARSA ligation and subclavian to carotid transposition, followed by thoracic endovascular aortic repair. Conclusions: Patients with CoA should be carefully studied, considering the possible coexistence of other congenital aortic arch defects, such as ARSA. Hybrid repair is a safe and effective approach for this condition.
Msx1 haploinsufficiency modifies the Pax9-deficient cardiovascular phenotype.
Successful embryogenesis relies on the coordinated interaction between genes and tissues. The transcription factors Pax9 and Msx1 genetically interact during mouse craniofacial morphogenesis, and mice deficient for either gene display abnormal tooth and palate development. Pax9 is expressed specifically in the pharyngeal endoderm at mid-embryogenesis, and mice deficient for Pax9 on a C57Bl/6 genetic background also have cardiovascular defects affecting the outflow tract and aortic arch arteries giving double-outlet right ventricle, absent common carotid arteries and interruption of the aortic arch. In this study we have investigated both the effect of a different genetic background and Msx1 haploinsufficiency on the presentation of the Pax9-deficient cardiovascular phenotype. Compared to mice on a C57Bl/6 background, congenic CD1-Pax9-/- mice displayed a significantly reduced incidence of outflow tract defects but aortic arch defects were unchanged. Pax9-/- mice with Msx1 haploinsufficiency, however, have a reduced incidence of interrupted aortic arch, but more cases with cervical origins of the right subclavian artery and aortic arch, than seen in Pax9-/- mice. This alteration in arch artery defects was accompanied by a rescue in third pharyngeal arch neural crest cell migration and smooth muscle cell coverage of the third pharyngeal arch arteries. Although this change in phenotype could theoretically be compatible with post-natal survival, using tissue-specific inactivation of Pax9 to maintain correct palate development whilst inducing the cardiovascular defects was unable to prevent postnatal death in the mutant mice. Hyoid bone and thyroid cartilage formation were abnormal in Pax9-/- mice. Msx1 haploinsufficiency mitigates the arch artery defects in Pax9-/- mice, potentially by maintaining the survival of the 3rd arch artery through unimpaired migration of neural crest cells to the third pharyngeal arches. With the neural crest cell derived hyoid bone and thyroid cartilage also being defective in Pax9-/- mice, we speculate that the pharyngeal endoderm is a key signalling centre that impacts on neural crest cell behaviour highlighting the ability of cells in different tissues to act synergistically or antagonistically during embryo development.
Congenital heart defects in CHARGE: The molecular role of CHD7 and effects on cardiac phenotype and clinical outcomes.
CHARGE syndrome is characterized by a pattern of congenital anomalies (Coloboma of the eye, Heart defects, Atresia of the choanae, Retardation of growth, Genital abnormalities, and Ear abnormalities). De novo mutations of chromodomain helicase DNA binding protein 7 (CHD7) are the primary cause of CHARGE syndrome. The clinical phenotype is highly variable including a wide spectrum of congenital heart defects. Here, we review the range of congenital heart defects and the molecular effects of CHD7 on cardiovascular development that lead to an over-representation of atrioventricular septal, conotruncal, and aortic arch defects in CHARGE syndrome. Further, we review the overlap of cardiovascular and noncardiovascular comorbidities present in CHARGE and their impact on the peri-operative morbidity and mortality in individuals with CHARGE syndrome.
Publicações recentes
Comprehensive defect detection in mouse embryos and the heart by combining automated phenotyping with novel population averages and atlases.
🥇 Revisão sistemáticaMaternal Genotype and Dietary Vitamin A Modify Aortic Arch Phenotypes in a Mouse Model of 22q11DS.
📖 RevisãoMsx1 haploinsufficiency modifies the Pax9-deficient cardiovascular phenotype.
Hybrid Procedure for Coexistence of Coarctation of the Aorta and Aberrant Right Subclavian Artery: A Case Report and Review of the Literature.
Congenital heart defects in CHARGE: The molecular role of CHD7 and effects on cardiac phenotype and clinical outcomes.
📚 EuropePMC1 artigos no totalmostrando 8
Comprehensive defect detection in mouse embryos and the heart by combining automated phenotyping with novel population averages and atlases.
Differentiation; research in biological diversityMaternal Genotype and Dietary Vitamin A Modify Aortic Arch Phenotypes in a Mouse Model of 22q11DS.
International journal of molecular sciencesMsx1 haploinsufficiency modifies the Pax9-deficient cardiovascular phenotype.
BMC developmental biologyHybrid Procedure for Coexistence of Coarctation of the Aorta and Aberrant Right Subclavian Artery: A Case Report and Review of the Literature.
Vascular and endovascular surgeryCongenital heart defects in CHARGE: The molecular role of CHD7 and effects on cardiac phenotype and clinical outcomes.
American journal of medical genetics. Part C, Seminars in medical geneticsMaternal Overweight and Obesity and Risk of Congenital Heart Defects.
Journal of the American College of CardiologyEmbryogenesis and Adult Life in the Absence of Intrinsic Apoptosis Effectors BAX, BAK, and BOK.
CellRoss Procedure in Neonates and Infants: A European Multicenter Experience.
The Annals of thoracic surgeryAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Anomalia do arco aórtico.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Anomalia do arco aórtico
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Comprehensive defect detection in mouse embryos and the heart by combining automated phenotyping with novel population averages and atlases.
- Maternal Genotype and Dietary Vitamin A Modify Aortic Arch Phenotypes in a Mouse Model of 22q11DS.
- Hybrid Procedure for Coexistence of Coarctation of the Aorta and Aberrant Right Subclavian Artery: A Case Report and Review of the Literature.
- Msx1 haploinsufficiency modifies the Pax9-deficient cardiovascular phenotype.
- Congenital heart defects in CHARGE: The molecular role of CHD7 and effects on cardiac phenotype and clinical outcomes.American journal of medical genetics. Part C, Seminars in medical genetics· 2020· PMID 31833191mais citado
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1132(Orphanet)
- MONDO:0015236(MONDO)
- GARD:741(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55785350(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Anomalia do arco aórtico
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos (literatura)
- fonte: Orphanet