A Síndrome do Coloboma Renal (SCR) é uma doença genética que se caracteriza por problemas no desenvolvimento do nervo óptico e por malformação e desenvolvimento insuficiente dos rins.
Introdução
O que você precisa saber de cara
A Síndrome do Coloboma Renal (SCR) é uma doença genética que se caracteriza por problemas no desenvolvimento do nervo óptico e por malformação e desenvolvimento insuficiente dos rins.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 13 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 50 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisTranscription factor that may have a role in kidney cell differentiation (PubMed:24676634). Has a critical role in the development of the urogenital tract, the eyes, and the CNS
Nucleus
Papillorenal syndrome
An autosomal dominant disorder characterized by both ocular and renal anomalies, but may also include vesicoureteral reflux, high frequency hearing loss, central nervous system anomalies, and/or genital anomalies. Eye anomalies in this disorder consist of a wide and sometimes excavated dysplastic optic disk with the emergence of the retinal vessels from the periphery of the disk, designated optic nerve coloboma or 'morning glory' anomaly. Associated findings may include a small corneal diameter, retinal coloboma, scleral staphyloma, optic nerve cyst, microphthalmia, and pigmentary macular dysplasia. The kidneys are small and abnormally formed (renal hypodysplasia), and have fewer than the normal number of glomeruli, which are enlarged (oligomeganephronia). These ocular and renal anomalies result in decreased visual acuity and retinal detachment, as well as hypertension, proteinuria, and renal insufficiency that frequently progresses to end-stage renal disease.
Variantes genéticas (ClinVar)
523 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 426 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
3 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome rim coloboma
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
Epigenetic, Genetic, and Functional Germline Alterations of PAX Genes in Human Pathology: A Comprehensive Update.
Paired box (PAX) genes encode a family of nine transcription factors that function as master regulators of embryogenesis, organogenesis, and lineage specification. Their tightly regulated spatial and temporal expression is essential for the development of multiple organ systems, including the central nervous system, eyes, kidneys, immune system, musculoskeletal system, and endocrine organs. Germline mutations of PAX genes result in a broad and often pleiotropic spectrum of human disease, reflecting the developmental programs governed by each family member. Pathogenic variants in PAX genes underlie diverse congenital disorders such as aniridia (PAX6), renal coloboma syndrome (PAX2), otofaciocervical syndrome with immunodeficiency (PAX1), Waardenburg syndrome (PAX3), maturity-onset diabetes of the young (PAX4), and tooth agenesis (PAX9). These conditions frequently demonstrate variable expressivity, incomplete penetrance, and overlapping phenotypes, which make it challenging to be clinically recognized. Beyond embryogenesis and embryologic development, emerging evidence indicates that several PAX proteins remain active in postnatal tissue maintenance, adult stem cell regulation, immune function, and regenerative responses (particularly PAX7 in skeletal muscle satellite cells and PAX5 in B-cell homeostasis), further expanding their clinical relevance. This review provides a synopsis of the major, clinically relevant, germline PAX gene mutations, emphasizing genotype-phenotype correlations, developmental mechanisms, and disease classification across the organ systems. By integrating molecular genetics with human pathology, we highlight the diagnostic implications of PAX genes as central determinants of congenital disease and provide a framework for understanding how alterations in the developmental transcriptional networks translate into human pathology.
Expanded Phenotype of PAX2-Related Papillorenal Syndrome: A Case Featuring FSGS, Atypical Retinopathy, Cerebellar Hypoplasia, and ADHD.
Papillorenal syndrome (PAPRS), or renal coloboma syndrome, is a rare autosomal dominant disorder caused by PAX2 mutations. It classically manifests with renal hypodysplasia and optic nerve anomalies. However, recent literature suggests an expanding phenotypic spectrum. We report a 7-year-8-month-old boy born to consanguineous parents, presenting with stage 4 chronic kidney disease (CKD), nephrotic-range proteinuria, visual impairment, and ADHD. Renal biopsy revealed focal segmental glomerulosclerosis (FSGS), and ocular examination showed bilateral peripheral scalloped chorioretinal atrophy without optic nerve colobomas. Genetic testing confirmed a pathogenic heterozygous PAX2 frameshift mutation (c.69_70insG; p.Val26fs28*), establishing the diagnosis of PAPRS. This case illustrates an expanded phenotype of PAX2-related PAPRS, including FSGS, atypical retinal degeneration, cerebellar hypoplasia, and ADHD. Recognition of such atypical presentations is vital for early diagnosis and multidisciplinary management, especially in resource-limited settings where classic features may be absent.
Genotype of PAX2-related disorders correlates with kidney and ocular manifestations.
PAX2-related disorders encompass renal coloboma syndrome (RCS) and hereditary focal segmental glomerulosclerosis (FSGS) type 7. We retrospectively analyzed 27 Korean patients with PAX2 pathogenic variants detected between 2004 and 2022 and conducted a literature review of 328 cases, including 301 previously reported. In our cohort, 19 had RCS, 4 had FSGS, and 4 had isolated congenital anomalies of the kidneys and urinary tract. Patients were classified by variant type into predicted loss of function (pLoF) and non-pLoF variant groups, and by variant location into paired domain and other sites group. pLoF variants were predominantly associated with RCS, observed in 82% of patients in both our data (18 of 22, P = 0.017) and the literature (140 of 171, P < 0.001). Kidney failure developed in 52% of Korean patients at a median age of 14.5 years, with no difference in kidney survival between variant types. However, the literature review indicated faster progression to kidney failure in patients with pLoF variants (11.0 vs. 24.0 years; pLoF, n = 138 vs. non-pLoF, n = 71; P = 0.002), with no significant difference by variant location. Ocular manifestations were more common, had earlier onset, and were more severe in the pLoF variants group in our cohort (P = 0.038). The literature confirmed a higher prevalence of ocular involvement in patients with pLoF variants (pLoF, n = 175 vs. non-pLoF, n = 88; P < 0.001) and in those with paired domain variants (P = 0.01). pLoF variants in PAX2 were associated with worse kidney and ocular outcomes. These findings support genotype-phenotype correlations, contributing to tailored management in patients with PAX2-related disorders.
'No causative variants found': an unusual presentation of PAX2-related disorder not detected on rapid whole exome sequencing testing.
Paired box 2 (PAX2)-related disorder, also known as renal coloboma syndrome, is a variably penetrant autosomal dominant condition, associated with renal and ophthalmological abnormalities. We report a child with PAX2-related disorder who presented atypically with acute ataxia on a background of stage 3 chronic kidney disease. Extensive biochemical, radiological and gene agnostic rapid trio exome sequencing was non-diagnostic. Identification of bilateral optic disc colobomas in the proband and his father raised the suspicion of an inherited PAX2-related disorder. No causative variants were identified on a focused review of the filtered genomic data. Given the strong suspicion of an inherited monogenic disorder, whole genome trio sequencing was requested. Analysis assuming incomplete penetrance identified a paternally inherited PAX2 microdeletion encompassing exon 4. This case adds to evidence of a broader PAX2-associated phenotype. It highlights the importance of a clinical genetics and mainstream interface when navigating and interpreting genetic testing.
C3 Glomerulonephritis in a Child with Renal Coloboma Syndrome.
Publicações recentes
A Case Report of Renal Coloboma Syndrome.
Epigenetic, Genetic, and Functional Germline Alterations of PAX Genes in Human Pathology: A Comprehensive Update.
Expanded Phenotype of PAX2-Related Papillorenal Syndrome: A Case Featuring FSGS, Atypical Retinopathy, Cerebellar Hypoplasia, and ADHD.
Presentation of Patients With Congenital Anomalies of the Kidney and Urinary Tract and PAX2 Loss-of-Function Variants and Implications for Clinical Management.
C3 Glomerulonephritis in a Child with Renal Coloboma Syndrome.
📚 EuropePMC46 artigos no totalmostrando 43
Epigenetic, Genetic, and Functional Germline Alterations of PAX Genes in Human Pathology: A Comprehensive Update.
Current issues in molecular biologyExpanded Phenotype of PAX2-Related Papillorenal Syndrome: A Case Featuring FSGS, Atypical Retinopathy, Cerebellar Hypoplasia, and ADHD.
Clinical case reportsPresentation of Patients With Congenital Anomalies of the Kidney and Urinary Tract and PAX2 Loss-of-Function Variants and Implications for Clinical Management.
Kidney international reportsC3 Glomerulonephritis in a Child with Renal Coloboma Syndrome.
Indian journal of pediatricsOcular clues to a renal diagnosis: classic imaging in renal coloboma syndrome.
Journal of nephrologyChoroidal neovascularization complicating papillorenal (renal coloboma) syndrome.
Journal francais d'ophtalmologieVariable Phenotypic Expression of PAX2 Variants in Two Lithuanian Families with Kidney Disease.
Medicina (Kaunas, Lithuania)Clinical spectrum, genetics and management insights of PAX2-related disorder in nine children.
European journal of medical researchGenotype of PAX2-related disorders correlates with kidney and ocular manifestations.
European journal of human genetics : EJHG'No causative variants found': an unusual presentation of PAX2-related disorder not detected on rapid whole exome sequencing testing.
BMJ case reportsFrameshift Mutation in PAX2 Related to Focal Segmental Glomerular Sclerosis: A Case Report and Literature Review.
Molecular genetics & genomic medicineRenal coloboma syndrome/dominant optic atrophy with severe retinal atrophy and de novo digenic mutations in PAX2 and OPA1.
Pediatric nephrology (Berlin, Germany)Familial focal segmental glomerulosclerosis with Alport-like glomerular basement changes caused by paired box protein 2 gene variant.
CEN case reportsPAX2 Gene Mutation in Pediatric Renal Disorders-A Narrative Review.
International journal of molecular sciencesRenal Coloboma Syndrome-An Autosomal Dominant Genetic Disorder.
The Indian journal of radiology & imagingPAX2/Renal Coloboma Syndrome Expresses Extreme Intrafamilial Phenotypic Variability.
NephronA case of renal coloboma syndrome.
Journal of nephrologyPAX2 Mutation-Related Renal Hypodysplasia: Review of the Literature and Three Case Reports.
Frontiers in pediatricsOcular phenotype in a patient with PAX2 gene mutation-associated papillorenal syndrome.
Ophthalmic geneticsPhenotypic spectrum and genetics of PAX2-related disorder in the Chinese cohort.
BMC medical genomicsResolving severe oligohydramnios as an early prenatal presentation of renal coloboma syndrome-A report of two generations.
Clinical case reportsUltra-rare renal diseases diagnosed with whole-exome sequencing: Utility in diagnosis and management.
BMC medical genomics[A family of renal coloboma syndrome].
[Zhonghua yan ke za zhi] Chinese journal of ophthalmologyMinor dysmorphic features in a patient with papillorenal syndrome: A Case Report.
JPMA. The Journal of the Pakistan Medical AssociationIdentification of candidate PAX2-regulated genes implicated in human kidney development.
Scientific reportsPAX2 variant associated with bilateral kidney agenesis and broad intrafamilial disease variability.
Clinical kidney journalPAX2 Mutation-Related Oligomeganephronia in a Young Adult Patient.
Case reports in nephrology and dialysisBilateral Morning Glory Anomaly With Optic Nerve Multiple Cysts.
Journal of neuro-ophthalmology : the official journal of the North American Neuro-Ophthalmology SocietyRenal coloboma syndrome with epilepsy.
The Korean journal of internal medicineWhole genome sequence analysis identifies a PAX2 mutation to establish a correct diagnosis for a syndromic form of hyperuricemia.
American journal of medical genetics. Part AClinical and genetic variability of PAX2-related disorder in the Japanese population.
Journal of human geneticsA novel truncating PAX2 mutation in a boy with renal coloboma syndrome with focal segmental glomerulosclerosis causing rapid progression to end-stage kidney disease.
CEN case reportsDiverse phenotypes in children with PAX2-related disorder.
Molecular genetics & genomic medicineDominant PAX2 mutations may cause steroid-resistant nephrotic syndrome and FSGS in children.
Pediatric nephrology (Berlin, Germany)New PAX2 heterozygous mutation in a child with chronic kidney disease: a case report and review of the literature.
BMC nephrologyDetection of copy number variations by pair analysis using next-generation sequencing data in inherited kidney diseases.
Clinical and experimental nephrologyThree New PAX2 Gene Mutations in Patients with Papillorenal Syndrome.
Neuro-ophthalmology (Aeolus Press)[Infrequent mutation in renal-coloboma syndrome: case report and review].
Archivos argentinos de pediatriaSEVERE CASE OF RENAL COLOBOMA SYNDROME IN LONG-TERM FOLLOW-UP.
Retinal cases & brief reportsPAX2 is dispensable for in vitro nephron formation from human induced pluripotent stem cells.
Scientific reportsComprehensive First-Line Magnetic Resonance Imaging in Hypertension: Experience From a Single-Center Tertiary Referral Clinic.
Journal of clinical hypertension (Greenwich, Conn.)Diverse Renal Phenotypes Observed in a Single Family with a Genetic Mutation in Paired Box Protein 2.
Case reports in nephrology and dialysisAssociation of PAX2 and Other Gene Mutations with the Clinical Manifestations of Renal Coloboma Syndrome.
PloS oneAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome rim coloboma.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome rim coloboma
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Epigenetic, Genetic, and Functional Germline Alterations of PAX Genes in Human Pathology: A Comprehensive Update.
- Expanded Phenotype of PAX2-Related Papillorenal Syndrome: A Case Featuring FSGS, Atypical Retinopathy, Cerebellar Hypoplasia, and ADHD.
- Genotype of PAX2-related disorders correlates with kidney and ocular manifestations.
- 'No causative variants found': an unusual presentation of PAX2-related disorder not detected on rapid whole exome sequencing testing.
- C3 Glomerulonephritis in a Child with Renal Coloboma Syndrome.
- A Case Report of Renal Coloboma Syndrome.
- Presentation of Patients With Congenital Anomalies of the Kidney and Urinary Tract and PAX2 Loss-of-Function Variants and Implications for Clinical Management.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1475(Orphanet)
- OMIM OMIM:120330(OMIM)
- MONDO:0007352(MONDO)
- GARD:4106(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q7133011(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
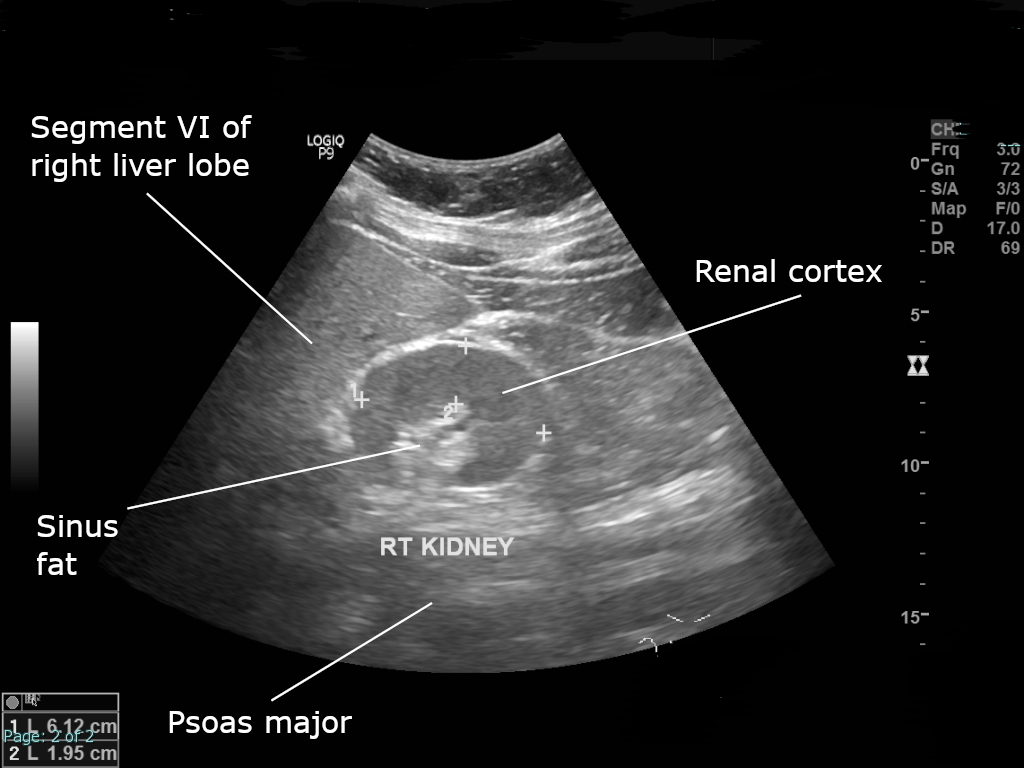
Síndrome rim coloboma
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata