Uma epilepsia generalizada de causa desconhecida, que se manifesta com o aparecimento de vários tipos de crises epilépticas nos primeiros anos de vida e está associada a um prognóstico desfavorável (uma condição que tende a ser grave). Pessoas afetadas apresentam regressão cognitiva (perda de habilidades mentais já adquiridas) e deficiência intelectual. A causa dessa condição é uma mutação (alteração) heterozigota (em apenas uma das duas cópias) no gene SLC6A1, localizado no cromossomo 3p25.
Introdução
O que você precisa saber de cara
Uma epilepsia generalizada de causa desconhecida, que se manifesta com o aparecimento de vários tipos de crises epilépticas nos primeiros anos de vida e está associada a um prognóstico desfavorável (uma condição que tende a ser grave). Pessoas afetadas apresentam regressão cognitiva (perda de habilidades mentais já adquiridas) e deficiência intelectual. A causa dessa condição é uma mutação (alteração) heterozigota (em apenas uma das duas cópias) no gene SLC6A1, localizado no cromossomo 3p25.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 17 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 51 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
7 genes identificados com associação a esta condição. Padrão de herança: Unknown.
Curadoria gene-doença
fontes oficiaisATP-dependent chromatin-remodeling factor that specifically binds to the promoter of target genes, leading to chromatin remodeling, possibly by promoting deposition of histone H3.3. Involved in myogenesis via interaction with MYOD1: binds to myogenic gene regulatory sequences and mediates incorporation of histone H3.3 prior to the onset of myogenic gene expression, promoting their expression (By similarity)
Nucleus
Developmental and epileptic encephalopathy 94
A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE94 is an autosomal dominant, severe form characterized by onset of multiple seizure types in the first few years of life.
Major constituent of the PSD essential for postsynaptic signaling. Inhibitory regulator of the Ras-cAMP pathway. Member of the NMDAR signaling complex in excitatory synapses, it may play a role in NMDAR-dependent control of AMPAR potentiation, AMPAR membrane trafficking and synaptic plasticity. Regulates AMPAR-mediated miniature excitatory postsynaptic currents. Exhibits dual GTPase-activating specificity for Ras and Rap. May be involved in certain forms of brain injury, leading to long-term lea
Intellectual developmental disorder, autosomal dominant 5
A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD5 patients show global developmental delay with delayed motor development, hypotonia, moderate-to-severe intellectual disability, and severe language impairment. Epilepsy and autism can be present in some patients.
Facilitative glucose transporter, which is responsible for constitutive or basal glucose uptake (PubMed:10227690, PubMed:10954735, PubMed:18245775, PubMed:19449892, PubMed:25982116, PubMed:27078104, PubMed:32860739). Has a very broad substrate specificity; can transport a wide range of aldoses including both pentoses and hexoses (PubMed:18245775, PubMed:19449892). Most important energy carrier of the brain: present at the blood-brain barrier and assures the energy-independent, facilitative trans
Cell membraneMelanosomePhotoreceptor inner segment
GLUT1 deficiency syndrome 1
A neurologic disorder showing wide phenotypic variability. The most severe 'classic' phenotype comprises infantile-onset epileptic encephalopathy associated with delayed development, acquired microcephaly, motor incoordination, and spasticity. Onset of seizures, usually characterized by apneic episodes, staring spells, and episodic eye movements, occurs within the first 4 months of life. Other paroxysmal findings include intermittent ataxia, confusion, lethargy, sleep disturbance, and headache. Varying degrees of cognitive impairment can occur, ranging from learning disabilities to severe intellectual disability.
Pore-forming subunit of Nav1.1, a voltage-gated sodium (Nav) channel that directly mediates the depolarizing phase of action potentials in excitable membranes. Navs, also called VGSCs (voltage-gated sodium channels) or VDSCs (voltage-dependent sodium channels), operate by switching between closed and open conformations depending on the voltage difference across the membrane. In the open conformation they allow Na(+) ions to selectively pass through the pore, along their electrochemical gradient.
Cell membrane
Generalized epilepsy with febrile seizures plus 2
A rare autosomal dominant, familial condition with incomplete penetrance and large intrafamilial variability. Patients display febrile seizures persisting sometimes beyond the age of 6 years and/or a variety of afebrile seizure types. This disease combines febrile seizures, generalized seizures often precipitated by fever at age 6 years or more, and partial seizures, with a variable degree of severity.
Involved in neurite outgrowth by regulating cell-cell adhesion via the N-cadherin signaling pathway. May act by regulating expression of protein-coding genes, such as N-cadherins and integrin beta-1 (ITGB1)
NucleusCytoplasm
Intellectual developmental disorder, X-linked 98
A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. XLID98 patients show delayed psychomotor development, absent or poor speech development, and postnatal growth retardation, often with microcephaly. Some patients show autistic behavioral features, such as stereotypic hand movements and repetitive behaviors. Additional, more variable features include spasticity, axial hypotonia, seizures, drooling, gastroesophageal reflux, and lack of sphincter control.
Component of the adaptor protein complex 2 (AP-2) (PubMed:12694563, PubMed:12952941, PubMed:14745134, PubMed:14985334, PubMed:15473838, PubMed:31104773). Adaptor protein complexes function in protein transport via transport vesicles in different membrane traffic pathways (PubMed:12694563, PubMed:12952941, PubMed:14745134, PubMed:14985334, PubMed:15473838, PubMed:31104773). Adaptor protein complexes are vesicle coat components and appear to be involved in cargo selection and vesicle formation (Pu
Cell membraneMembrane, coated pit
Intellectual developmental disorder, autosomal dominant 60, with seizures
An autosomal dominant disorder characterized by global developmental delay apparent in the first six months of life, followed by onset of seizures between 21 months and 4 years. Disease features include moderate-to-severe intellectual disability, poor speech, delayed walking, and ataxia.
Mediates transport of gamma-aminobutyric acid (GABA) together with sodium and chloride and is responsible for the reuptake of GABA from the synapse (PubMed:30132828). The translocation of GABA, however, may also occur in the reverse direction leading to the release of GABA (By similarity). The direction and magnitude of GABA transport is a consequence of the prevailing thermodynamic conditions, determined by membrane potential and the intracellular and extracellular concentrations of Na(+), Cl(-
Cell membranePresynapse
Myoclonic-atonic epilepsy
A form of epilepsy characterized by myoclonic-atonic and absence seizures, appearing in early childhood. Patients have delayed development before the onset of seizures and show varying degrees of intellectual disability following seizure onset.
Variantes genéticas (ClinVar)
1.849 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 811 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
28 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Epilepsia com crises mioclônico-atônicas
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
From clinical practice to mechanistic insights in ketogenic diets for epilepsy.
Ketogenic diet therapies, including the classic ketogenic diet, modified Atkins diet, and low glycaemic index treatment, have shown effectiveness in controlling seizures, in part by shifting metabolism from glucose to ketone bodies. They improve mitochondrial function, reduce neuroinflammation, and modulate neurotransmitters. Ketogenic diet therapies also affect the gut microbiome, potentially impacting neurotransmitter balance in ways that contribute to seizure control. A classic ketogenic diet is effective yet restrictive, whereas the modified Atkins diet and low glycaemic index treatment offer greater flexibility, tolerability, and ease of implementation, particularly in resource-limited settings. Cochrane reviews and meta-analyses rank the certainty of randomised controlled trial evidence for ketogenic diet therapies as limited. Early initiation of ketogenic diet therapies, particularly in children or patients with metabolic epilepsies, improves seizure outcomes, potentially preventing further mitochondrial and neuronal damage and reducing the risk of developing resistance to antiseizure medications. Research using rigorous, large-scale comparative effectiveness study designs that accounts for differences in age, epilepsy type, dietary therapy modality, sociodemographic background, care delivery contexts, and that minimises performance and observation bias is needed to resolve remaining uncertainties regarding the efficacy and real-world challenges of ketogenic diet therapies in epilepsy.
Epilepsy Phenotypic Spectrum of NUS1-Related Disorder: A Case Series.
Epilepsy with myoclonic and atonic seizures (EMAtS), also known as Doose syndrome, accounts for 1%-2% of childhood epilepsies, and various genes have been implicated in causing this epilepsy syndrome. NUS1 encodes for Nogo-B receptor (NgBR), which stabilizes the dehydrodolichyl-diphosphate synthase complex in the endoplasmic reticulum, promoting its enzymatic activity (cis-IPTase) and thereby regulating cholesterol biosynthesis. Pathogenic variants in NUS1 have been associated with movement disorder and epilepsy; however, the spectrum of epilepsy and electroencephalogram (EEG) phenotype has not been well characterized. We describe a single-center case series of five patients with NUS1-related disorder. In our cohort, three patients met the diagnostic criteria of EMAtS, and the remainder had a milder form of generalized epilepsy. Four patients had a pathogenic variant in NUS1 on one allele, and one patient had a missense change of unclear significance but fit the phenotype of NUS1-related disorder. All patients bearing the pathogenic variants in NUS1 had normal to mild developmental delay at the onset of epilepsy, with normal brain magnetic resonance imaging. Age of seizure onset in these patients was 1-7 years, and patients responded to levetiracetam and/or valproic acid. The EEG findings for these patients included the presence of spike and slow wave discharges, as well as the presence of generalized, invariant monomorphic theta range activity in the awake state, which was seen in four out of the five patients. Taken together, NUS1 variants are associated with generalized epilepsy phenotype and an invariant EEG pattern of monomorphic theta activity.
Epilepsy with myoclonic-atonic seizures: genetic aetiologies, outcomes and prognostic indicators.
Epilepsy with myoclonic-atonic seizures, formerly myoclonic-astatic epilepsy or Doose syndrome, accounts for 1-2.2% of childhood-onset epilepsies. We investigated genetic determinants, long-term clinical outcomes and prognostic indicators in a large cohort using homogeneous inclusion criteria. We studied 60 patients (26.7% female), mean age 14.5 years (±9.1, range 3.2-41), followed between 1986 and 2024 at two paediatric neurology centres. Average follow-up was 11.7 years. Inclusion criteria were seizure onset between 6 months and 8 years, generalized 2-6 Hz spike-wave discharges and video-EEG documented myoclonic-atonic, myoclonic seizures or both. We analysed clinical, EEG, neuroimaging, neuropsychological and genetic data obtained with next-generation sequencing. We used χ² test, t-test, Log-rank test, Cox regression, population-averaged logistic models and Benjamini-Yekutieli procedure to identify predictors of seizure outcome, intellectual disability and other neurodevelopmental comorbidities. We observed myoclonic-atonic seizures in 55/60 (91.7%), tonic-vibratory seizures in 44/60 (73.4%), absence seizures in 30/60 (50%), myoclonic seizures without post-myoclonic atonia in 25/60 (42%) and non-convulsive status epilepticus in 13/60 (21.7%). A 'stormy' onset occurred in 26/60 patients (43.3%). The most effective drugs were valproate, ethosuximide, benzodiazepines and phenobarbital, used in different combinations, whereas the newer drugs offered no benefit. Long-term outcomes were variable. Thirty-seven patients (61.7%) achieved seizure freedom after 5.1 years on average. We observed drug resistance in 23/60 patients (38.3%) and intellectual disability in 35/60 (58.3%). One adult patient died (mortality rate 1.80/1000-person-years). Attention deficit hyperactivity disorder was the most common comorbidity (24/60, 40%). 'Stormy' onset did not predict a worse prognosis. Global developmental delay at epilepsy onset was associated with drug resistance (P = 0.004, Q = 0.064) and with intellectual disability (P = 0.003, Q = 0.048). We found pathogenic variants in 15/39 (38.5%) patients undergoing next-generation sequencing, including four genes novel for this syndrome (KMT2E; POGZ; SHANK3; YWHAG), with exome sequencing yielding higher diagnostic rates than gene panels. Epilepsy with myoclonic-atonic seizures is a complex syndrome with diverse genetic causes and variable seizure severity and outcomes. Our findings expand its genetic landscape and highlight the prognostic value of prompt overall neurodevelopmental assessment at clinical onset. Whole exome sequencing should be prioritized for early diagnosis and counselling.
The Spike-Hyperslow-Wave (SHsW) complex: a distinctive ictal EEG hallmark of myoclonic-atonic seizures in EMAtS.
Focus on epilepsy and epilepsy syndromes in children with autism spectrum disorders: a study of 74 patients.
Epilepsy is a common finding in children with autism spectrum disorders (ASD), but few studies describe the characteristics of epilepsy in these children. Our study aimed to characterize the electroclinical features of children with ASD and epilepsy through a retrospective multicenter study. Patients with ASD who subsequently developed epilepsy seen at nine pediatric neurology departments were included. Patients with developmental and epileptic encephalopathies (DEE), chronic neurological diseases with epilepsy who developed autism, and those with non-epileptic paroxysmal disorders were excluded. Overall, 74 patients were included, accounting for 15 % of 494 children with ASD seen between 2015 and 2023; 39 were female (52.7 %) and 35 male (47.3 %). Focal epilepsies were identified in 43 patients (58.1 %), which were non-self-limited in 24 and self-limited in 19. Generalized epilepsies were observed in 19 (25.7 %), including six with generalized tonic-clonic seizures alone (one in childhood, five in adolescence), nine with juvenile myoclonic epilepsy, one with childhood absence epilepsy, and three with juvenile absence epilepsy. Eight patients (10.8 %) had epileptic encephalopathies: EE-SWAS in six and epilepsy with myoclonic atonic seizures in two. Four patients (5.4 %) had combined focal and generalized epilepsy. No significant differences were found between epilepsy syndrome or type of epilepsy, seizure type, and comorbidities. No specific epilepsy phenotype was identified in our patients with ASD; the types of epilepsy and syndromes were similar to those seen in the general population. Management should address both epilepsy and the broader complexities of ASD through an integrated approach.
Publicações recentes
Epilepsy Phenotypic Spectrum of NUS1-Related Disorder: A Case Series.
The Spike-Hyperslow-Wave (SHsW) complex: a distinctive ictal EEG hallmark of myoclonic-atonic seizures in EMAtS.
Epilepsy with myoclonic-atonic seizures: genetic aetiologies, outcomes and prognostic indicators.
Compound heterozygous variants of CACNA1H change channel properties and contribute to intractable epilepsy with myoclonic-atonic seizures.
📖 RevisãoCognitive and behavioral performance in children with epilepsy with myoclonic-atonic seizures.
📖 Revisão📚 EuropePMC32 artigos no totalmostrando 65
From clinical practice to mechanistic insights in ketogenic diets for epilepsy.
The Lancet. NeurologyEpilepsy Phenotypic Spectrum of NUS1-Related Disorder: A Case Series.
Annals of the Child Neurology SocietyThe Spike-Hyperslow-Wave (SHsW) complex: a distinctive ictal EEG hallmark of myoclonic-atonic seizures in EMAtS.
Epilepsy & behavior : E&BEpilepsy with myoclonic-atonic seizures: genetic aetiologies, outcomes and prognostic indicators.
Brain communicationsCompound heterozygous variants of CACNA1H change channel properties and contribute to intractable epilepsy with myoclonic-atonic seizures.
Journal of human geneticsCognitive and behavioral performance in children with epilepsy with myoclonic-atonic seizures.
Dementia & neuropsychologiaFocus on epilepsy and epilepsy syndromes in children with autism spectrum disorders: a study of 74 patients.
Brain & developmentA new developmental and epileptic encephalopathy: PUM1-neurodevelopmental disorder with epilepsy with myoclonic-atonic seizures.
SeizureGenetic etiologies with a large NGS panel in a monocentric cohort of 1000 patients with pediatric onset epilepsies.
Epilepsia openCenobamate in developmental and epileptic encephalopathies and generalized epilepsies: A case report on epilepsy with myoclonic-atonic seizures and systematic review of current evidence.
SeizureEpilepsy with myoclonic-atonic seizures: an update on genetic causes, nosological limits, and treatment strategies.
The Lancet. NeurologyImpact of vagus nerve stimulation on refractory epilepsy with myoclonic atonic seizures: Case series insights.
SeizureClinical and genetic analysis of epilepsy with myoclonic-atonic seizures caused by SLC6A1 gene variant.
Frontiers in pediatricsFelbamate as a therapeutic alternative to drug-resistant genetic generalized epilepsy: a systematic review and meta-analysis.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyKetogenic diet therapy for the treatment of pediatric epilepsy.
Epileptic disorders : international epilepsy journal with videotapeEvaluating the patient needs and tolerability of Clobazam liquid formulation (Likozam® 1 mg/mL): A French patient and care-givers' centered survey.
Epilepsy & behavior : E&BRapid seizure resolution with cannabidiol in medically refractory epilepsy with myoclonic-atonic seizures.
Epileptic disorders : international epilepsy journal with videotapePerampanel reduces seizure frequency in patients with developmental and epileptic encephalopathy for a long term.
Scientific reportsPOLR3B is associated with a developmental and epileptic encephalopathy with myoclonic-atonic seizures and ataxia.
EpilepsiaA generalized seizure type: Myoclonic-to-tonic seizure.
Clinical neurophysiology : official journal of the International Federation of Clinical NeurophysiologyPopulation-based study of rare epilepsy incidence in a US urban population.
EpilepsiaHaploinsufficiency underlies the neurodevelopmental consequences of SLC6A1 variants.
American journal of human geneticsPredictors of genetic diagnosis in individuals with developmental and epileptic encephalopathies.
Epilepsy & behavior : E&BComprehensive phenotypes of patients with SYNGAP1-related disorder reveals high rates of epilepsy and autism.
EpilepsiaEfficacy of felbamate in a cohort of patients with epilepsy with myoclonic atonic seizures (EMAtS).
Epilepsy researchRefractory tonic-myoclonic status epilepticus with catamenial recurrence in epilepsy with myoclonic atonic seizures: A case report.
HeliyonCreating rare epilepsy cohorts using keyword search in electronic health records.
EpilepsiaSleep and respiratory abnormalities in adults with developmental and epileptic encephalopathies using polysomnography and video-EEG monitoring.
Epilepsia openCannabidiol in children with treatment-resistant epilepsy with myoclonic-atonic seizures.
Epilepsy & behavior : E&BEfficacy of a pre-specified timeline-based treatment protocol in children with acute repetitive seizures or seizure clusters.
Journal of neurosciences in rural practiceA case of epilepsy with myoclonic atonic seizures caused by SLC6A1 gene mutation due to balanced chromosomal translocation.
Brain & developmentEpidemiology of Developmental and Epileptic Encephalopathy and of Intellectual Disability and Epilepsy in Children.
NeurologyMED13 mutation: A novel cause of developmental and epileptic encephalopathy with infantile spasms.
SeizureInternational League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE Task Force on Nosology and Definitions.
EpilepsiaLong-term outcome of developmental and epileptic encephalopathies.
Revue neurologiqueEpilepsy with myoclonic-atonic seizures, also known as Doose syndrome: Modification of the diagnostic criteria.
European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology SocietyGABRB3-related epilepsy: novel variants, clinical features and therapeutic implications.
Journal of neurologyStructural brain abnormalities in epilepsy with myoclonic atonic seizures.
Epilepsy researchIctal vocalizations are relatively common in myoclonic-atonic seizures associated with Doose syndrome: an audio-video-polygraphic analysis.
Epileptic disorders : international epilepsy journal with videotapeEpilepsy With Myoclonic Atonic Seizures: Why Is the Yield of Genetic Testing for a "Presumed Genetic" Epilepsy Low?
Epilepsy currentsMorphometric analysis of spike-wave complexes (SWCs) causing myoclonic seizures in children with idiopathic myoclonic epilepsies - A positive SWC component correlates with myoclonic intensity.
Brain & developmentResults of an international Delphi consensus in epilepsy with myoclonic atonic seizures/ Doose syndrome.
SeizureCurrent knowledge of SLC6A1-related neurodevelopmental disorders.
Brain communicationsEpilepsy with myoclonic-atonic seizures (Doose syndrome): Clarification of diagnosis and treatment options through a large retrospective multicenter cohort.
EpilepsiaClinical and genetic characteristics of patients with Doose syndrome.
Epilepsia openPhenotypic and genetic spectrum of epilepsy with myoclonic atonic seizures.
EpilepsiaCHD2-related epilepsy: novel mutations and new phenotypes.
Developmental medicine and child neurologyDissecting the phenotypic and genetic spectrum of early childhood-onset generalized epilepsies.
SeizureAdolescent-onset absence epilepsy years after resolution of childhood epilepsy with myoclonic-atonic seizures.
Epilepsy & behavior reportsA Recurrent Missense Variant in AP2M1 Impairs Clathrin-Mediated Endocytosis and Causes Developmental and Epileptic Encephalopathy.
American journal of human geneticsHow often is antiseizure drug-free ketogenic diet therapy achieved?
Epilepsy & behavior : E&BGene mutations in paediatric epilepsies cause NMDA-pathy, and phasic and tonic GABA-pathy.
Developmental medicine and child neurologyGenetic testing in a cohort of patients with potential epilepsy with myoclonic-atonic seizures.
Epilepsy researchDiagnosis switching and outcomes in a cohort of patients with potential epilepsy with myoclonic-atonic seizures.
Epilepsy researchHow do we diagnose and treat epilepsy with myoclonic-atonic seizures (Doose syndrome)? Results of the Pediatric Epilepsy Research Consortium survey.
Epilepsy researchSuccessful corpus callosotomy for Doose syndrome.
Brain & developmentEpilepsy with myoclonic atonic seizures and chronic cerebellar symptoms associated with antibodies against glutamate receptors N2B and D2 in serum and cerebrospinal fluid.
Epileptic disorders : international epilepsy journal with videotapeMutations in GABRB3: From febrile seizures to epileptic encephalopathies.
NeurologySLC6A1 Mutation and Ketogenic Diet in Epilepsy With Myoclonic-Atonic Seizures.
Pediatric neurologyEpileptic spasms in epilepsy with myoclonic-atonic seizures (Doose syndrome).
Epileptic disorders : international epilepsy journal with videotapeEpilepsy with myoclonic-atonic seizures (Doose syndrome): When video-EEG polygraphy holds the key to syndrome diagnosis.
Epilepsy & behavior case reportsMultiplex families with epilepsy: Success of clinical and molecular genetic characterization.
NeurologyCHD2 mutations are a rare cause of generalized epilepsy with myoclonic-atonic seizures.
Epilepsy & behavior : E&BThree siblings with multiform seizures: An unusual presentation of Doose syndrome.
The National medical journal of IndiaMutations in the GABA Transporter SLC6A1 Cause Epilepsy with Myoclonic-Atonic Seizures.
American journal of human geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Epilepsia com crises mioclônico-atônicas.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Epilepsia com crises mioclônico-atônicas
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- From clinical practice to mechanistic insights in ketogenic diets for epilepsy.
- Epilepsy Phenotypic Spectrum of NUS1-Related Disorder: A Case Series.
- Epilepsy with myoclonic-atonic seizures: genetic aetiologies, outcomes and prognostic indicators.
- The Spike-Hyperslow-Wave (SHsW) complex: a distinctive ictal EEG hallmark of myoclonic-atonic seizures in EMAtS.
- Focus on epilepsy and epilepsy syndromes in children with autism spectrum disorders: a study of 74 patients.
- Compound heterozygous variants of CACNA1H change channel properties and contribute to intractable epilepsy with myoclonic-atonic seizures.
- Cognitive and behavioral performance in children with epilepsy with myoclonic-atonic seizures.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1942(Orphanet)
- OMIM OMIM:616421(OMIM)
- MONDO:0014633(MONDO)
- Epilepsia(PCDT · Ministério da Saúde)
- GARD:16108(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
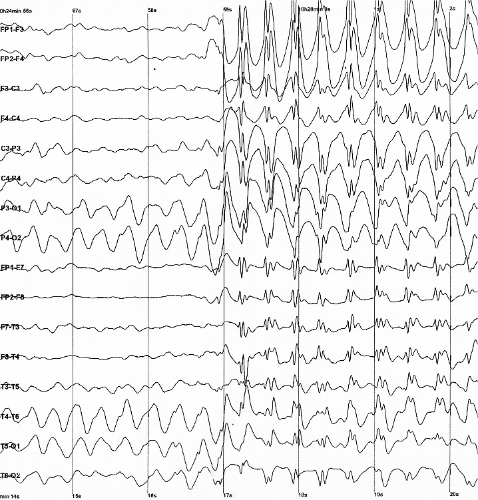
Epilepsia com crises mioclônico-atônicas
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Reposicionamento
- fonte: Drug Repurposing Hub