A oligomeganefronia é uma condição séria de desenvolvimento dos rins, sendo a forma mais grave de hipoplasia renal — ou seja, quando os rins não se formam completamente. Ela se caracteriza por uma redução de 80% no número de néfrons (as unidades que filtram o sangue nos rins), e por um aumento acentuado (hipertrofia) no tamanho dos glomérulos e túbulos, que são partes importantes desses néfrons.
Introdução
O que você precisa saber de cara
A oligomeganefronia é uma condição séria de desenvolvimento dos rins, sendo a forma mais grave de hipoplasia renal — ou seja, quando os rins não se formam completamente. Ela se caracteriza por uma redução de 80% no número de néfrons (as unidades que filtram o sangue nos rins), e por um aumento acentuado (hipertrofia) no tamanho dos glomérulos e túbulos, que são partes importantes desses néfrons.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 7 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 30 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Oligomeganefronia
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Bilateral Glomerulocystic Kidney Disease With Extensive Embryonal Hyperplasia in a Setting of HNF1B Mutation.
Glomerulocystic renal disease has numerous etiologies, including HNF1B mutations. In addition to cysts, morphologic renal findings in a setting of HNF1B mutations include cystic renal dysplasia, solitary functioning kidney, horseshoe kidney, and oligomeganephronia. Embryonal hyperplasia resembling nephrogenic rests has been reported in rare cases of glomerulocystic disease, but none have been genetically characterized. We report a case of bilateral glomerulocystic kidney disease (GCKD) showing extensive embryonal hyperplasia in a setting of germline HNF1B mutation with progressive renal failure. Explant showed numerous epithelial proliferations throughout the intervening stroma. GCKD may be seen in a setting of HNF1B mutation; however, the additional finding of extensive embryonal hyperplasia in a case with a known mutation has never been reported. Reports of similar embryonal hyperplasia, associated with either cystic kidney disease or other disease processes, appear to represent a heterogeneous population of presentations and etiologies, though there is sufficient evidence to suggest that this is a finding, that is, recurrently associated with cystic kidney diseases. The underlying pathobiology of embryonal hyperplasia and the neoplastic potential in this setting is unknown. We report this case to highlight a novel combination of morphologic and genetic findings in GCKD and to raise awareness of this rare finding.
Renal Hypoplasia and Oligomeganephronia in a Fetus with Wolf-Hirschhorn Syndrome.
Background and Clinical Significance: Wolf-Hirschhorn syndrome (WHS, OMIM #194190) is caused by deletion of the distal short arm of chromosome 4. It is characterized by intrauterine growth restriction (IUGR), developmental delay, epilepsy, distinctive facial features, and urinary tract anomalies, particularly renal hypoplasia. However, the histological profile of renal involvement in WHS is rarely documented. Case presentation: We report a case of fetal WHS with renal hypoplasia and histological evidence of oligomeganephronia (OMN). At 21 weeks' gestation, a prenatal ultrasound revealed oligo/anhydramnios and IUGR. Genetic testing (karyotype and CGH-array) confirmed a de novo 17.92 Mb terminal deletion from 4p16.3 to 4p15.31. The pregnancy was legally terminated at 23 weeks. The autopsy showed characteristic WHS dysmorphisms, growth restriction, and markedly small kidneys. Histology revealed OMN with a thinned renal cortex with reduced glomeruli, mainly hypoplastic, some of which were hypertrophic, and dilated proximal tubules. Scattered medullary tubules were present within the tubulointerstitial compartment, alongside thickened tubular basement membranes highlighted by Collagen IV staining. Conclusions: This case suggests that OMN may be a histological hallmark of renal hypoplasia in WHS, especially in larger 4p deletions. Recognizing this pattern may help with prenatal prognosis and clinical management. Further studies are needed to confirm this association.
Oligomeganephronia with PAX2 gene deletion diagnosed at the third renal biopsy: a case report.
Oligomeganephronia is a congenital anomaly of the kidney and urinary tract. It is often categorized as one of the hypoplastic kidney conditions. The pathological diagnosis of oligomeganephronia is challenged by the absence of clear diagnostic criteria, which often leads to subjective interpretations by pathologists. This report presents the case of a 7-year-old girl who was diagnosed with oligomeganephronia through a third renal biopsy, which was confirmed by gene analysis revealing PAX2 deletion. Two previous renal biopsies, with the naked eye through a microscope, failed to identify glomerular hypertrophy and sparse glomerular distribution density. However, using digital images, the glomeruli were larger than those of age-matched controls, and the number of glomeruli within the renal cortex area revealed sparse glomerular distribution density. Image processing allows for objective evaluation of the glomerular size and glomerular distribution density, providing a quantitative assessment. For earlier diagnosis of oligomeganephronia, an appropriate objective standardized method for measuring glomerular size and glomerular distribution density should be established.
Phenotype-Genotype Correlations in Three Different Cases of Adult-Onset Genetic Focal Segmental Glomerulosclerosis.
This study highlights the importance of a combined diagnostic approach in the diagnosis of rare diseases, such as adult-onset genetic FSGS. We present three adult patient cases evaluated with kidney biopsy for proteinuria, chronic kidney disease, and hypertension, which were suggestive of adult-onset genetic FSGS. Renal biopsy samples and formalin-fixed, paraffin-embedded fetal kidneys were evaluated using standard light microscopical stainings, direct immunofluorescence on cryostat sections, and electron microscopy. Clinical exome sequencing was performed for each case, and 45 FSGS-related genes were analyzed. Identifying mutations in the PAX2, ACTN4, and COL4A5 genes have prompted a re-evaluation of the previous histopathological examinations. The PAX2 mutation led to a thinner nephrogenic zone and decreased number of glomeruli, resulting in oligohydramnios during fetal development and oligomeganephronia and adaptive focal-segmental glomerulosclerosis in adulthood. The ACTN4 mutation caused distinct electron-dense aggregates in podocyte cell bodies, while the COL4A5 mutation led to segmental sclerosis of glomeruli with marked interstitial fibrosis and tubular atrophy. The identification of specific mutations and their histopathological consequences can lead to a better understanding of the disease and its progression, as well as potential treatment options.
A novel pathogenic variant c.262delA in PBX1 causing oligomeganephronia identified using whole-exome sequencing and a literature review.
Oligomeganephronia (OMN) is a rare congenital renal hypoplasia reported more often in children than in adults. The diagnosis of OMN relies on renal biopsy and exhibits a significant reduction in the number of glomeruli and pronounced glomerular hypertrophy. Here, we report the case of an 8-year-old boy with recurrent proteinuria and abnormal external ears. A renal biopsy revealed large and rare glomeruli. The histological findings confirmed the diagnosis of OMN. Whole-exome sequencing of the patient revealed a new pathogenic variant in PBX1 (hg19, NM_002585, c.262delA, p.Thr88Glnfs*3). The PBX1 gene encodes a transcription factor whose pathogenic variants can result in congenital renal and urinary system anomalies, with or without hearing loss, abnormal ears, and developmental retardation (CAKUTED). This is the first report to detect PBX1 pathogenic variants in children with OMN, a novel phenotype of human PBX1 pathogenic variants. We performed functional prediction analyses of deletions in the corresponding structural domains. We summarized 27 cases of PBX1 single pathogenic variants reported between 2003 and 2023 in terms of truncating and missense pathogenic variants, which can deepen our understanding of the PBX1 structural domain and expand our knowledge of the PBX1 genotype and phenotype.
Publicações recentes
Bilateral Glomerulocystic Kidney Disease With Extensive Embryonal Hyperplasia in a Setting of HNF1B Mutation.
Renal Hypoplasia and Oligomeganephronia in a Fetus with Wolf-Hirschhorn Syndrome.
Phenotype-Genotype Correlations in Three Different Cases of Adult-Onset Genetic Focal Segmental Glomerulosclerosis.
Oligomeganephronia with PAX2 gene deletion diagnosed at the third renal biopsy: a case report.
A novel pathogenic variant c.262delA in PBX1 causing oligomeganephronia identified using whole-exome sequencing and a literature review.
📚 EuropePMC39 artigos no totalmostrando 17
Bilateral Glomerulocystic Kidney Disease With Extensive Embryonal Hyperplasia in a Setting of HNF1B Mutation.
Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology SocietyRenal Hypoplasia and Oligomeganephronia in a Fetus with Wolf-Hirschhorn Syndrome.
Diagnostics (Basel, Switzerland)Phenotype-Genotype Correlations in Three Different Cases of Adult-Onset Genetic Focal Segmental Glomerulosclerosis.
International journal of molecular sciencesOligomeganephronia with PAX2 gene deletion diagnosed at the third renal biopsy: a case report.
Journal of nephrologyA novel pathogenic variant c.262delA in PBX1 causing oligomeganephronia identified using whole-exome sequencing and a literature review.
American journal of medical genetics. Part AProteinuria and Renal Dysfunction Due to Extremely Low Birth Weight in a Patient with Silver-Russell Syndrome.
The Kurume medical journalPathological and clinical characteristics of late-onset oligomeganephronia based on a histomorphometric study.
BMC nephrologyClinical and pathological investigation of oligomeganephronia.
Pediatric nephrology (Berlin, Germany)A Case Report and Literature Review of Oligomeganephronia.
Frontiers in medicinePAX2 Mutation-Related Oligomeganephronia in a Young Adult Patient.
Case reports in nephrology and dialysisGlomerular galactose-deficient IgA1 expression analysis in pediatric patients with glomerular diseases.
Scientific reportsRenal Hypoplasia, From Grossly Insufficient to Not Quite Enough: Consideration for Expanded Concepts Based Upon the Author's Perspective With Historical Review.
Advances in anatomic pathologyTGFBI-associated corneal dystrophy and nephropathy: a novel syndrome?
CEN case reportsOligonephronia and Wolf-Hirschhorn syndrome: A further observation.
American journal of medical genetics. Part AOsteogenesis imperfecta complicated with renal hypoplasia leads to chronic kidney disease.
Pediatrics international : official journal of the Japan Pediatric SocietyHeterozygous p.S811F RET gene mutation associated with renal agenesis, oligomeganephronia and total colonic aganglionosis: a case report.
BMC nephrologyOligomeganephronia: case report and literature review.
Srpski arhiv za celokupno lekarstvoAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Oligomeganefronia.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Oligomeganefronia
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Bilateral Glomerulocystic Kidney Disease With Extensive Embryonal Hyperplasia in a Setting of HNF1B Mutation.Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society· 2026· PMID 41731920mais citado
- Renal Hypoplasia and Oligomeganephronia in a Fetus with Wolf-Hirschhorn Syndrome.
- Oligomeganephronia with PAX2 gene deletion diagnosed at the third renal biopsy: a case report.
- Phenotype-Genotype Correlations in Three Different Cases of Adult-Onset Genetic Focal Segmental Glomerulosclerosis.
- A novel pathogenic variant c.262delA in PBX1 causing oligomeganephronia identified using whole-exome sequencing and a literature review.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2260(Orphanet)
- MONDO:0016407(MONDO)
- GARD:4066(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Q50349808(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
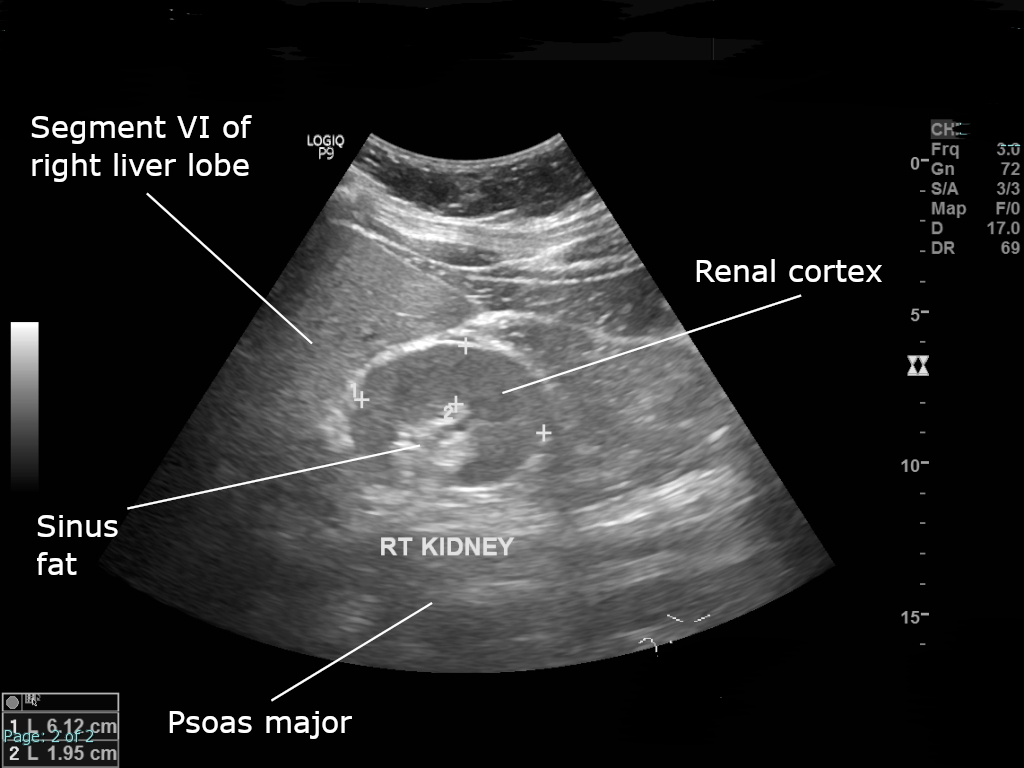
Oligomeganefronia
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata