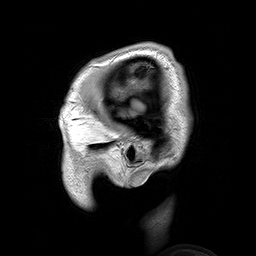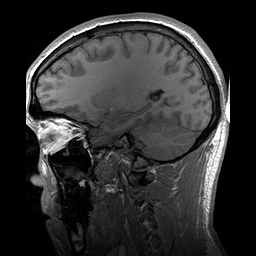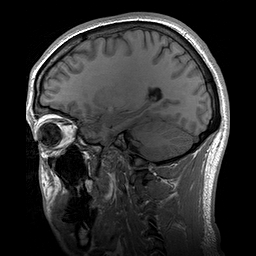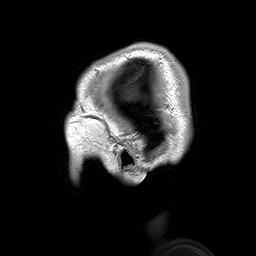
A esclerosteose é uma síndrome de hiperostose esclerosante grave muito rara, caracterizada clinicamente por sindactilia variável e crescimento excessivo do esqueleto progressivo (particularmente do crânio), resultando em características faciais distintas (crescimento excessivo da mandíbula, protuberância frontal, hipoplasia médio-facial), compressão de nervos cranianos causando paralisia facial e surdez, e elevação potencialmente letal da pressão intracraniana.
Introdução
O que você precisa saber de cara
A esclerosteose é uma síndrome de hiperostose esclerosante grave muito rara, caracterizada clinicamente por sindactilia variável e crescimento excessivo do esqueleto progressivo (particularmente do crânio), resultando em características faciais distintas (crescimento excessivo da mandíbula, protuberância frontal, hipoplasia médio-facial), compressão de nervos cranianos causando paralisia facial e surdez, e elevação potencialmente letal da pressão intracraniana.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 20 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 61 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Mediates SOST-dependent inhibition of bone formation. Functions as a specific facilitator of SOST-mediated inhibition of Wnt signaling. Plays a key role in the formation and the maintenance of the neuromuscular junction (NMJ), the synapse between motor neuron and skeletal muscle. Directly binds AGRIN and recruits it to the MUSK signaling complex. Mediates the AGRIN-induced phosphorylation of MUSK, the kinase of the complex. The activation of MUSK in myotubes induces the formation of NMJ by regul
Cell membrane
Cenani-Lenz syndactyly syndrome
A congenital malformation syndrome defined as complete and complex syndactyly of the hands combined with malformations of the forearm bones and similar manifestations in the lower limbs. It is characterized by fusion and disorganization of metacarpal and phalangeal bones, radius and ulnar shortening, radioulnar synostosis, and severe syndactyly of hands and feet.
Negative regulator of bone growth that acts through inhibition of Wnt signaling and bone formation
Secreted, extracellular space, extracellular matrix
Sclerosteosis 1
An autosomal recessive sclerosing bone dysplasia characterized by a generalized hyperostosis and sclerosis leading to a markedly thickened skull, with mandible, ribs, clavicles and all long bones also being affected. Due to narrowing of the foramina of the cranial nerves, facial nerve palsy, hearing loss and atrophy of the optic nerves can occur. Sclerosteosis is clinically and radiologically very similar to van Buchem disease, mainly differentiated by hand malformations and a large stature in sclerosteosis patients.
Variantes genéticas (ClinVar)
126 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 1.265 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
3 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Esclerosteose
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Clinical Characteristics and Management of Rare Metabolic Bone Diseases: An Audit of the Rare Metabolic Bone Disease Registry of India.
Rare diseases, defined by the 2002 Rare Disease Act, affect fewer than 5 in 10,000 individuals. Rare metabolic bone diseases (MBDs), such as osteogenesis imperfecta, hypophosphatasia, osteopetrosis, and other unclassified disorders, can disrupt bone development and remodeling, posing diagnostic and management challenges. This study analyzed data from the rarembd.in registry (2010-2024), a 15-year database documenting only rare MBDs. Clinical presentation and demographic data of patients with rare MBDs were collated. Common MBDs (osteoporosis, primary hyperparathyroidism) were excluded. Genetic testing was performed in a subset of patients. There was a total of 218 patients with an almost equal gender distribution (male-to-female ratio of 1:1.07) and a mean age of 29.1 ± 18.9 years. The registry identified 29 rare MBDs with three main disease categories: demineralization disorders (50.4%), disorders of bone matrix and cartilage formation (32.5%), and sclerotic disorders (13.7%); with a smaller proportion categorized as unclassified bone disorders (2.7%). Rickets/osteomalacia (27.1%) was the most common, followed by osteogenesis imperfecta (23.4%) and fibrous dysplasia/McCune-Albright syndrome (18.8%). Fractures affected 57.7% of patients, with 24.5% experiencing multiple fractures, while 31.1% exhibited skeletal deformities. Mutation analysis in our registry identified pathogenic variants in the SOST, TGFβ1, SLC34A3, ALPL, and VCP genes, confirming the genetic basis of sclerosteosis, Camurati-Engelmann disease, hypophosphatemic rickets, hypophosphatasia, and IBMPFD, respectively. Different management strategies were used that included teriparatide, bisphosphonates (zoledronate or alendronate) with total contact casting, intralesional zoledronate, denosumab, calcium, active vitamin D, and recombinant human growth hormone. Total parathyroidectomy was performed in specific cases. The registry classified RMBDs into four categories, with demineralization disorders being the most common, followed by bone matrix/cartilage formation disorders, sclerotic diseases, and unclassified cases. There were 29 RMBDs, and rickets/osteomalacia was the most prevalent subtype, tumor-induced osteomalacia followed by familial hypophosphatemic osteomalacia. Among the unclassified bone disorders, fragility fractures emerged as the most common presentation.
Novel Loss of Function Variant in SOST From Chinese Family Results in Sclerosteosis 1.
SOST encodes a secreted glycoprotein that is similar in sequence to the differential screening-selected gene aberrative in neuroblastoma (DAN) family of bone morphogenetic protein (BMP) antagonists. Pathogenic variants in the SOST gene result in sclerosteosis, van Buchem disease (VBD), or craniodiaphyseal dysplasia. SOST-related genetic disorders are very rare, and limited studies have reported variants associated with sclerosteosis. Clinical tests such as magnetic resonance imaging (MRI), computed tomography (CT), emission computed tomography (ECT), electromyogram (EMG), routine blood tests, and physical examinations were conducted for the proband. Trio-whole exome sequencing (Trio-WES) was performed, and the rare variants (allele frequency < 0.01) in the exon and splicing regions were selected for further pathogenic evaluation. Candidate pathogenic variants were validated through Sanger sequencing. The wild and mutant SOST sequences were cloned into the pcDNA3.1 expression vector, and the RNA and protein expression levels were investigated in the HEK293T cell line. In this study, we present a case study of a proband who displays abnormal facial expressions accompanied by numbness. The results of the brain MRI show thickening of the skull and disappearance of the diplopia signal. The temporal bone CT scan indicates diffuse osteosclerosis affecting the bilateral ossicular chains and internal auditory meatus, as well as stenosis of the bilateral internal auditory meatus. Trio-WES sequencing detected a novel homozygous variant in the proband: NM_025237.3(SOST): c.327C>A (p.Cys109*), which was also validated in his sister from the same family. According to the ACMG guidelines, the variant is classified as "likely pathogenic." The in vitro experiments demonstrated that the variant caused a decrease in SOST expression at RNA and protein level and produced a truncated protein. The report presents new evidence for the clinical diagnosis of SOST-related facial numbness and expands the variant spectrum of SOST.
Insights into Natural History, Phenotypic, and Molecular Spectrum in a Large Cohort of Osteosclerotic Disorders.
Osteosclerotic bone diseases include more than 30 rare diseases characterized by excessive bone formation. The aim of this study is to compare the molecular pathogenesis and prognostic features of 12 different osteosclerotic diseases. Thirty-four patients from 23 families were included, 25 of whom were followed for a period of one to 22 years. Exome sequencing was performed in 20 families. Primary hypertrophic osteoarthropathy (PHOAR1/2) was found in 12 patients, followed by juvenile Paget's disease (JPD)-5 in five, craniometaphyseal dysplasia (CMD) and Camurati-Engelmann disease (CED) in four, Ghosal hematodiaphyseal dysplasia (GHDD) in three patients, sclerosteosis-1 in two patients, and ultra-rare diseases including trichothiodystrophy-1, prenatal Caffey disease, melorheosteosis, and Lenz-Majewski hyperostotic dwarfism in one patient each. Patients with CMD and sclerosteosis-1 had severe cranial sclerosis leading to facial dysmorphism. CMD was characterized by metaphyseal widening, radiolucency, and diaphyseal sclerosis of the long bones in early childhood and later developed Erlenmeyer flask deformity sparing the vertebrae and pelvis, whereas sclerosteosis-1 manifested as generalized sclerosis. CED and GHDD share bone pain, difficulty in walking, and diaphyseal sclerosis, with some patients also having bone marrow involvement. Interestingly, patients with CED and JPD-5 showed osteopenia in early childhood, followed by the development of osteosclerosis in late childhood. Clinical and radiologic findings improved over time in PHOAR1 patients, whereas they progressed in JPD-5 and trichothiodystrophy-1 patients. Intra- and interfamilial clinical differences were observed in CMD, CED, JPD-5, and GHDD. The knowledge gained about the natural history of osteosclerotic diseases will make an important contribution to their diagnosis and management.
Porcupine inhibition is a promising pharmacological treatment for severe sclerosteosis pathologies.
Sclerosteosis, an ultra-rare disorder characterised by high bone mass (HBM) and skeletal overgrowth, leads to facial paralysis, hearing loss and raised intracranial pressure, which is currently managed only through high-risk surgery. Sclerosteosis is caused by SOST mutations and loss of functional sclerostin, a protein that suppresses osteogenesis by antagonising Wnt/β-catenin signalling. Herein, using in vitro and in vivo approaches, we explore whether LGK974, another potent Wnt inhibitor that targets porcupine (PORCN, Wnt-specific acyltransferase), is a promising sclerosteosis therapeutic. In vitro assays showed that 100 nmol/L LGK974 significantly reduced osteoblast alkaline phosphatase (ALP) activity/mineralisation, decreased Wnt/osteoblast marker (Axin2, Runx2 and Ocn) expression, and downregulated ossification and the Wnt signalling pathway, without affecting osteoclast numbers/resorption. To assess in vivo effects, 6-week-old male and female Sost deficient (Sost-/-) mice received LGK974 for 4 weeks and right hindlimbs were subjected to 20 N peak loading to assess mechanoadaptive interactions. µCT revealed significant reductions in vertebral trabecular number and lower cortical bone volume in loaded and non-loaded tibiae in male and female LGK974-treated Sost-/- mice. Interestingly, the target engagement biomarker Axin2 was only significantly reduced in male vertebrae, which may indicate differences in male and female response to LGK974. This study also shows that PORCN inhibition may effectively limit characteristic HBM and skeletal overgrowth in sclerosteosis patients at sites with severe pathology.
Long-term clinical and bone mineral density changes of adult patients with sclerostin deficiency due to van Buchem disease: a follow-up study.
Van Buchem disease (VBD) is an inherited rare sclerosing bone disorder, due to defective synthesis of sclerostin, a negative regulator of bone formation. Our earlier cross-sectional studies of patients with VBD and the closely related sclerosteosis suggested that the accrual of bone mass does not continue after puberty but longitudinal studies of patients with sclerostin deficiency are not available. The aim, therefore, of the present study was the long-term assessment of adult patients with VBD. Fifteen previously evaluated patients with genetically confirmed VBD were invited to participate in the study and 11 (4 women) consented. Mean follow-up time was 8.9 ± 1.1 yr and median age at follow-up was 47 yr (range 20-60). Seven patients developed permanent facial paresis, 9 had progressing hearing loss, and 2 developed had increased intracranial pressure requiring cranial surgery. Dental problems were common, and 3 patients developed osteoarthritis during follow-up. None experienced a cardiovascular event. BMD did not change at the LS or the left FN; Z-scores were 10.2 ± 1.3 SD vs 9.4 ± 2.3 SD, p=.62, and 8.9 ± 2.2 SD vs 7.7 ± 2.2 SD, p=.15, respectively. The variability of the clinical expression and progression of the disease, despite the stabilization of BMD but with progressive cranial nerve compression, requires continuous monitoring of these patients for whom no disease-specific therapy is currently available.
Publicações recentes
Clinical Characteristics and Management of Rare Metabolic Bone Diseases: An Audit of the Rare Metabolic Bone Disease Registry of India.
Trapped cranial nerve and dense bones-sclerosteosis due to LRP4 mutation.
Novel Loss of Function Variant in SOST From Chinese Family Results in Sclerosteosis 1.
Insights into Natural History, Phenotypic, and Molecular Spectrum in a Large Cohort of Osteosclerotic Disorders.
Porcupine inhibition is a promising pharmacological treatment for severe sclerosteosis pathologies.
📚 EuropePMC59 artigos no totalmostrando 67
Clinical Characteristics and Management of Rare Metabolic Bone Diseases: An Audit of the Rare Metabolic Bone Disease Registry of India.
Calcified tissue internationalTrapped cranial nerve and dense bones-Sclerosteosis due to LRP4 mutation.
QJM : monthly journal of the Association of PhysiciansNovel Loss of Function Variant in SOST From Chinese Family Results in Sclerosteosis 1.
Molecular genetics & genomic medicineInsights into Natural History, Phenotypic, and Molecular Spectrum in a Large Cohort of Osteosclerotic Disorders.
Calcified tissue internationalPorcupine inhibition is a promising pharmacological treatment for severe sclerosteosis pathologies.
Bone researchLong-term clinical and bone mineral density changes of adult patients with sclerostin deficiency due to van Buchem disease: a follow-up study.
JBMR plusTherapeutic targeting of Wnt antagonists by small molecules for treatment of osteoporosis.
Biochemical pharmacologyLRP4 site-specific variants in the third β-propeller domain causes congenital myasthenic syndrome type 17.
European journal of medical geneticsAn Additional Lrp4 High Bone Mass Mutation Mitigates the Sost-Knockout Phenotype in Mice by Increasing Bone Remodeling.
Calcified tissue internationalMef2c regulates bone mass through Sost-dependent and -independent mechanisms.
BoneNew mutation in the β1 propeller domain of LRP4 responsible for congenital myasthenic syndrome associated with Cenani-Lenz syndrome.
Scientific reportsSclerostin: clinical insights in muscle-bone crosstalk.
The Journal of international medical researchInhibiting WNT secretion reduces high bone mass caused by Sost loss-of-function or gain-of-function mutations in Lrp5.
Bone researchSostdc1 Suppression in the Absence of Sclerostin Potentiates Anabolic Action of Cortical Bone in Mice.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchExamining craniofacial variation among crispant and mutant zebrafish models of human skeletal diseases.
Journal of anatomyInvestigation of the Underlying Mechanism of Sclerosteosis Expression in Muscle Tissue in Multiple Myeloma with Sarcopenia.
Journal of inflammation researchNovel insights on the effect of sclerostin on bone and other organs.
The Journal of endocrinologyNeurological manifestations in patients and disease carriers in an Italian family with osteosclerosis.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologySclerostin ablation prevents aortic valve stenosis in mice.
American journal of physiology. Heart and circulatory physiologyPositive and Negative Regulators of Sclerostin Expression.
International journal of molecular sciencesEvidence for the major contribution of remodeling-based bone formation in sclerostin-deficient mice.
BoneA Novel Mutation in a Gene Causes Sclerosteosis in a Family of Mediterranean Origin.
Medicina (Kaunas, Lithuania)Identification of Compound Heterozygous Variants in LRP4 Demonstrates That a Pathogenic Variant outside the Third β-Propeller Domain Can Cause Sclerosteosis.
GenesRecombinant sclerostin inhibits bone formation in vitro and in a mouse model of sclerosteosis.
Journal of orthopaedic translationDifferential diagnosis of a diffuse sclerosis in an identified male skull (early 20th century Coimbra, Portugal): A multimethodological approach for the identification of osteosclerotic dysplasias in skeletonized individuals.
International journal of paleopathologyLow magnitude vibration alleviates age-related bone loss by inhibiting cell senescence of osteogenic cells in naturally senescent rats.
AgingOpportunities and Challenges in Functional Genomics Research in Osteoporosis: Report From a Workshop Held by the Causes Working Group of the Osteoporosis and Bone Research Academy of the Royal Osteoporosis Society on October 5th 2020.
Frontiers in endocrinologyCortical bone adaptation to a moderate level of mechanical loading in male Sost deficient mice.
Scientific reportsLooking for new anabolic treatment from rare diseases of bone formation.
The Journal of endocrinologySclerostin: from bench to bedside.
Journal of bone and mineral metabolismGenomic Medicine: Lessons Learned From Monogenic and Complex Bone Disorders.
Frontiers in endocrinologyMolecular Basis for Craniofacial Phenotypes Caused by Sclerostin Deletion.
Journal of dental researchThe Osteocyte as the New Discovery of Therapeutic Options in Rare Bone Diseases.
Frontiers in endocrinologyTitanium particles damage osteocytes and inhibit osteoblast differentiation.
Journal of experimental orthopaedicsA novel biallelic splice-site variant in the LRP4 gene causes sclerosteosis 2.
Birth defects researchSOST Deficiency Aggravates Osteoarthritis in Mice by Promoting Sclerosis of Subchondral Bone.
BioMed research internationalAnatomical similarity between the Sost-knockout mouse and sclerosteosis in humans.
Anatomical record (Hoboken, N.J. : 2007)Sclerostin Antibody-Induced Changes in Bone Mass Are Site Specific in Developing Crania.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchDeficiency of lrp4 in zebrafish and human LRP4 mutation induce aberrant activation of Jagged-Notch signaling in fin and limb development.
Cellular and molecular life sciences : CMLSSclerosteosis: Report of type 1 or 2 in three Indian Tamil families and literature review.
BoneSclerostin neutralization unleashes the osteoanabolic effects of Dkk1 inhibition.
JCI insightConditional Deletion of Sost in MSC-Derived Lineages Identifies Specific Cell-Type Contributions to Bone Mass and B-Cell Development.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchMutations in the fourth β-propeller domain of LRP4 are associated with isolated syndactyly with fusion of the third and fourth fingers.
Human mutationA Review of the Clinical, Radiological and Biochemical Characteristics and Genetic Causes of High Bone Mass Disorders.
Current drug targetsSclerostin: Intracellular mechanisms of action and its role in the pathogenesis of skeletal and vascular disorders.
Advances in clinical and experimental medicine : official organ Wroclaw Medical UniversityGenetics of Sost/SOST in sclerosteosis and van Buchem disease animal models.
Metabolism: clinical and experimentalThe Lrp4R1170Q Homozygous Knock-In Mouse Recapitulates the Bone Phenotype of Sclerosteosis in Humans.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchSclerostin: From bedside to bench, and back to bedside.
BoneRole and mechanism of action of sclerostin in bone.
BoneSclerostin deficiency in humans.
BoneN-cadherin restrains PTH repressive effects on sclerostin/SOST by regulating LRP6-PTH1R interaction.
Annals of the New York Academy of SciencesSclerostin inhibition promotes TNF-dependent inflammatory joint destruction.
Science translational medicineThe genetics of bone mass and susceptibility to bone diseases.
Nature reviews. RheumatologyFrom disease to treatment: from rare skeletal disorders to treatments for osteoporosis.
EndocrineAnti-sclerostin - is there an indication?
InjuryA Novel Domain-Specific Mutation in a Sclerosteosis Patient Suggests a Role of LRP4 as an Anchor for Sclerostin in Human Bone.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchGenetic control of bone mass.
Molecular and cellular endocrinologySclerosteosis caused by a novel nonsense mutation of SOST in a consanguineous family.
Clinical geneticsMiscellaneous Bone Disorders.
Endocrine developmentLRP receptor family member associated bone disease.
Reviews in endocrine & metabolic disordersA Novel Loss-of-Sclerostin Function Mutation in a First Egyptian Family with Sclerosteosis.
BioMed research internationalNovel targets for the prevention of osteoporosis - lessons learned from studies of metabolic bone disorders.
Expert opinion on therapeutic targetsCrystallization and preliminary X-ray crystallographic analysis of the sclerostin-neutralizing Fab AbD09097.
Acta crystallographica. Section F, Structural biology communicationsSclerosteosis (craniotubular hyperostosis-syndactyly) with complex hyperphalangy of the index finger.
Pediatric radiologyLrp4 in osteoblasts suppresses bone formation and promotes osteoclastogenesis and bone resorption.
Proceedings of the National Academy of Sciences of the United States of AmericaSclerostin and skeletal health.
Reviews in endocrine & metabolic disordersThe sclerostin story: from human genetics to the development of novel anabolic treatment for osteoporosis.
Hormones (Athens, Greece)Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Esclerosteose.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Esclerosteose
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Clinical Characteristics and Management of Rare Metabolic Bone Diseases: An Audit of the Rare Metabolic Bone Disease Registry of India.
- Novel Loss of Function Variant in SOST From Chinese Family Results in Sclerosteosis 1.
- Insights into Natural History, Phenotypic, and Molecular Spectrum in a Large Cohort of Osteosclerotic Disorders.
- Porcupine inhibition is a promising pharmacological treatment for severe sclerosteosis pathologies.
- Long-term clinical and bone mineral density changes of adult patients with sclerostin deficiency due to van Buchem disease: a follow-up study.
- Trapped cranial nerve and dense bones-sclerosteosis due to LRP4 mutation.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:3152(Orphanet)
- MONDO:0017838(MONDO)
- GARD:4771(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q3042146(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Esclerosteose
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata