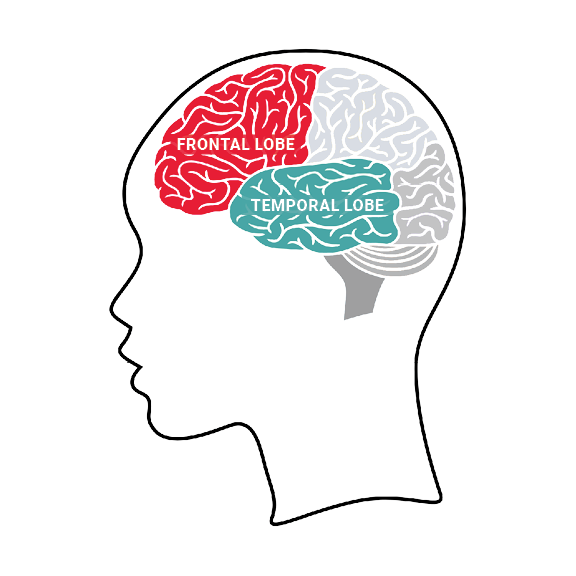
A Demência Frontotemporal com Doença do Neurônio Motor (DFT-DNM) é um tipo de desgaste nas regiões frontal e temporal do cérebro. Ela é caracterizada pelo início lento e gradual (geralmente entre os 38 e 78 anos) de sintomas psiquiátricos ligados à demência, como mudanças na personalidade, comportamento sem freios, irritabilidade e agressividade. Também causa dificuldades de memória, uma perda geral da capacidade intelectual, problemas emocionais e dificuldades de fala e comunicação (chamada afasia motora transcortical), que com o tempo podem levar a pessoa a ficar completamente sem conseguir falar (mutismo). Além disso, a doença apresenta manifestações da doença do neurônio motor, como o enfraquecimento e a perda de músculos (atrofia muscular neurogênica), semelhante ao que ocorre na Esclerose Lateral Amiotrófica (ELA). A doença piora progressivamente, e a morte ocorre geralmente entre 2 e 5 anos após o início dos sintomas.
Introdução
O que você precisa saber de cara
A Demência Frontotemporal com Doença do Neurônio Motor (DFT-DNM) é um tipo de desgaste nas regiões frontal e temporal do cérebro. Ela é caracterizada pelo início lento e gradual (geralmente entre os 38 e 78 anos) de sintomas psiquiátricos ligados à demência, como mudanças na personalidade, comportamento sem freios, irritabilidade e agressividade. Também causa dificuldades de memória, uma perda geral da capacidade intelectual, problemas emocionais e dificuldades de fala e comunicação (chamada afasia motora transcortical), que com o tempo podem levar a pessoa a ficar completamente sem conseguir falar (mutismo). Além disso, a doença apresenta manifestações da doença do neurônio motor, como o enfraquecimento e a perda de músculos (atrofia muscular neurogênica), semelhante ao que ocorre na Esclerose Lateral Amiotrófica (ELA). A doença piora progressivamente, e a morte ocorre geralmente entre 2 e 5 anos após o início dos sintomas.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 33 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 71 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
8 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant.
Molecular adapter required for selective macroautophagy (aggrephagy) by acting as a bridge between polyubiquitinated proteins and autophagosomes (PubMed:15340068, PubMed:15953362, PubMed:16286508, PubMed:17580304, PubMed:20168092, PubMed:22017874, PubMed:22622177, PubMed:24128730, PubMed:28404643, PubMed:29343546, PubMed:29507397, PubMed:31857589, PubMed:33509017, PubMed:34471133, PubMed:34893540, PubMed:35831301, PubMed:37306101, PubMed:37802024). Promotes the recruitment of ubiquitinated cargo
Cytoplasmic vesicle, autophagosomePreautophagosomal structureCytoplasm, cytosolNucleus, PML bodyLate endosomeLysosomeNucleusEndoplasmic reticulumCytoplasm, myofibril, sarcomere
Paget disease of bone 3
A disorder of bone remodeling characterized by increased bone turnover affecting one or more sites throughout the skeleton, primarily the axial skeleton. Osteoclastic overactivity followed by compensatory osteoblastic activity leads to a structurally disorganized mosaic of bone (woven bone), which is mechanically weaker, larger, less compact, more vascular, and more susceptible to fracture than normal adult lamellar bone.
Acts as a guanine-nucleotide releasing factor (GEF) for Rab GTPases by promoting the conversion of inactive RAB-GDP to the active form RAB-GTP (PubMed:27103069, PubMed:27193190, PubMed:27617292, PubMed:28195531, PubMed:37821429). Acts as a GEF for RAB39A which enables HOPS-mediated autophagosome-lysosome membrane tethering and fusion in mammalian autophagy (PubMed:37821429). Component of the C9orf72-SMCR8 complex where both subunits display GEF activity and that regulates autophagy (PubMed:27103
CytoplasmNucleusCytoplasm, P-bodyCytoplasm, Stress granuleEndosomeLysosomeCytoplasmic vesicle, autophagosomeAutolysosomeSecretedCell projection, axonCell projection, growth conePerikaryonCell projection, dendritePresynapsePostsynapseNucleus membrane
Frontotemporal dementia and/or amyotrophic lateral sclerosis 1
An autosomal dominant neurodegenerative disorder characterized by adult onset of frontotemporal dementia and/or amyotrophic lateral sclerosis in an affected individual. There is high intrafamilial variation. Frontotemporal dementia is characterized by frontal and temporal lobe atrophy associated with neuronal loss, gliosis, and dementia. Patients exhibit progressive changes in social, behavioral, and/or language function. Amyotrophic lateral sclerosis is characterized by the death of motor neurons in the brain, brainstem, and spinal cord, resulting in fatal paralysis.
RNA-binding protein that is involved in various steps of RNA biogenesis and processing (PubMed:23519609). Preferentially binds, via its two RNA recognition motifs RRM1 and RRM2, to GU-repeats on RNA molecules predominantly localized within long introns and in the 3'UTR of mRNAs (PubMed:23519609, PubMed:24240615, PubMed:24464995). In turn, regulates the splicing of many non-coding and protein-coding RNAs including proteins involved in neuronal survival, as well as mRNAs that encode proteins relev
NucleusCytoplasmCytoplasm, Stress granuleMitochondrion
Amyotrophic lateral sclerosis 10
A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases.
DNA/RNA-binding protein that plays a role in various cellular processes such as transcription regulation, RNA splicing, RNA transport, DNA repair and damage response (PubMed:27731383). Binds to ssRNA containing the consensus sequence 5'-AGGUAA-3' (PubMed:21256132). Binds to nascent pre-mRNAs and acts as a molecular mediator between RNA polymerase II and U1 small nuclear ribonucleoprotein thereby coupling transcription and splicing (PubMed:26124092). Also binds its own pre-mRNA and autoregulates
Nucleus
Serine/threonine kinase that plays an essential role in regulating inflammatory responses to foreign agents (PubMed:10581243, PubMed:11839743, PubMed:12692549, PubMed:12702806, PubMed:14703513, PubMed:15367631, PubMed:15485837, PubMed:18583960, PubMed:21138416, PubMed:23453971, PubMed:23453972, PubMed:23746807, PubMed:25636800, PubMed:26611359, PubMed:32404352, PubMed:34363755, PubMed:32298923). Following activation of toll-like receptors by viral or bacterial components, associates with TRAF3 a
Cytoplasm
Glaucoma 1, open angle, P
A form of primary open angle glaucoma (POAG). POAG is characterized by a specific pattern of optic nerve and visual field defects. The angle of the anterior chamber of the eye is open, and usually the intraocular pressure is increased. However, glaucoma can occur at any intraocular pressure. The disease is generally asymptomatic until the late stages, by which time significant and irreversible optic nerve damage has already taken place. GLC1P is characterized by early onset, thin central corneas and low intraocular pressure.
Necessary for the fragmentation of Golgi stacks during mitosis and for their reassembly after mitosis. Involved in the formation of the transitional endoplasmic reticulum (tER). The transfer of membranes from the endoplasmic reticulum to the Golgi apparatus occurs via 50-70 nm transition vesicles which derive from part-rough, part-smooth transitional elements of the endoplasmic reticulum (tER). Vesicle budding from the tER is an ATP-dependent process. The ternary complex containing UFD1, VCP and
Cytoplasm, cytosolEndoplasmic reticulumNucleusCytoplasm, Stress granule
Inclusion body myopathy with early-onset Paget disease with or without frontotemporal dementia 1
An autosomal dominant disease characterized by disabling muscle weakness clinically resembling to limb girdle muscular dystrophy, osteolytic bone lesions consistent with Paget disease, and premature frontotemporal dementia. Clinical features show incomplete penetrance.
Acts as a guanine-nucleotide releasing factor (GEF) for Rab GTPases by promoting the conversion of inactive RAB-GDP to the active form RAB-GTP (PubMed:27103069, PubMed:27193190, PubMed:27617292, PubMed:28195531, PubMed:37821429). Acts as a GEF for RAB39A which enables HOPS-mediated autophagosome-lysosome membrane tethering and fusion in mammalian autophagy (PubMed:37821429). Component of the C9orf72-SMCR8 complex where both subunits display GEF activity and that regulates autophagy (PubMed:27103
CytoplasmNucleusCytoplasm, P-bodyCytoplasm, Stress granuleEndosomeLysosomeCytoplasmic vesicle, autophagosomeAutolysosomeSecretedCell projection, axonCell projection, growth conePerikaryonCell projection, dendritePresynapsePostsynapseNucleus membrane
Frontotemporal dementia and/or amyotrophic lateral sclerosis 1
An autosomal dominant neurodegenerative disorder characterized by adult onset of frontotemporal dementia and/or amyotrophic lateral sclerosis in an affected individual. There is high intrafamilial variation. Frontotemporal dementia is characterized by frontal and temporal lobe atrophy associated with neuronal loss, gliosis, and dementia. Patients exhibit progressive changes in social, behavioral, and/or language function. Amyotrophic lateral sclerosis is characterized by the death of motor neurons in the brain, brainstem, and spinal cord, resulting in fatal paralysis.
May be involved in the maintenance of mitochondrial organization and mitochondrial cristae structure
Mitochondrion intermembrane space
Frontotemporal dementia and/or amyotrophic lateral sclerosis 2
A neurodegenerative disorder characterized by frontotemporal dementia and/or amyotrophic lateral sclerosis in affected individuals. There is high intrafamilial variation. Frontotemporal dementia is characterized by frontal and temporal lobe atrophy associated with neuronal loss, gliosis, and dementia. Patients exhibit progressive changes in social, behavioral, and/or language function. Amyotrophic lateral sclerosis is characterized by the death of motor neurons in the brain, brainstem, and spinal cord, resulting in fatal paralysis.
Variantes genéticas (ClinVar)
496 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
53 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Demência frontotemporal com doença do neurônio motor
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
14 ensaios clínicos encontrados, 3 ativos.
Publicações mais relevantes
Matrin-3 forms spherical and wormlike assemblies that are modulated by RNA binding and ALS/FTD-associated mutations.
Matrin-3 (MATR3) is an RNA-binding protein (RBP) that is associated with familial amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). MATR3 features two RNA recognition motifs, two zinc-finger motifs, and four intrinsically disordered regions. Here, we report that human MATR3 associates with itself to form nanoscale spherical assemblies at ultralow protein concentrations. Through concentration-dependent associations, the spheres, which are 20-30 nm in diameter, transition into wormlike assemblies. These observations are reminiscent of sphere-to-worm transitions and micellization of amphiphilic molecules. Using computations and experiments, we discovered that the pattern of inter-domain attractions and repulsions gives MATR3 an inverse bolaamphiphile-like architecture that explains the concentration-dependent assembly characteristics. RNA binding causes shortening of wormlike assemblies of MATR3, whereas ALS/FTD-associated mutations render MATR3 assemblies less responsive to modulation by RNA. Overall, our findings highlight the unique assemblies formed by MATR3 while also showing how RNA-dependent interactions and ALS/FTD-associated mutations modulate the assemblies.
A high-fidelity CRISPR-Cas13 system improves abnormalities associated with C9ORF72-linked ALS/FTD.
An abnormal expansion of a GGGGCC (G4C2) hexanucleotide repeat in the C9ORF72 gene is the most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), two debilitating neurodegenerative disorders driven in part by gain-of-function mechanisms involving transcribed forms of the repeat expansion. By utilizing a Cas13 variant with reduced collateral effects, we develop here a high-fidelity RNA-targeting CRISPR-based system for C9ORF72-linked ALS/FTD. When delivered to the brain of a transgenic rodent model, this Cas13-based platform curbed the expression of the G4C2 repeat-containing RNA without affecting normal C9ORF72 levels, which in turn decreased the formation of RNA foci, reduced the production of a dipeptide repeat protein, and reversed transcriptional deficits. This high-fidelity system possessed improved transcriptome-wide specificity compared to its native form and mediated targeting in motor neuron-like cells derived from a patient with ALS. These results lay the foundation for the implementation of RNA-targeting CRISPR technologies for C9ORF72-linked ALS/FTD.
Poly-GR repeats associated with ALS/FTD gene C9ORF72 impair translation elongation and induce a ribotoxic stress response in neurons.
Hexanucleotide repeat expansion in the C9ORF72 gene is the most frequent inherited cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). The expansion results in multiple dipeptide repeat proteins, among which arginine-rich poly-GR proteins are highly toxic to neurons and decrease the rate of protein synthesis. We investigated whether the effect on protein synthesis contributes to neuronal dysfunction and degeneration. We found that the expression of poly-GR proteins inhibited global translation by perturbing translation elongation. In iPSC-differentiated neurons, the translation of transcripts with relatively slow elongation rates was further slowed, and stalled, by poly-GR. Elongation stalling increased ribosome collisions and induced a ribotoxic stress response (RSR) mediated by ZAKα that increased the phosphorylation of the kinase p38 and promoted cell death. Knockdown of ZAKα or pharmacological inhibition of p38 ameliorated poly-GR-induced toxicity and improved the survival of iPSC-derived neurons from patients with C9ORF72-ALS/FTD. Our findings suggest that targeting the RSR may be neuroprotective in patients with ALS/FTD caused by repeat expansion in C9ORF72.
The exocyst subunit EXOC2 regulates the toxicity of expanded GGGGCC repeats in C9ORF72-ALS/FTD.
GGGGCC (G4C2) repeat expansion in C9ORF72 is the most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). How this genetic mutation leads to neurodegeneration remains largely unknown. Using CRISPR-Cas9 technology, we deleted EXOC2, which encodes an essential exocyst subunit, in induced pluripotent stem cells (iPSCs) derived from C9ORF72-ALS/FTD patients. These cells are viable owing to the presence of truncated EXOC2, suggesting that exocyst function is partially maintained. Several disease-relevant cellular phenotypes in C9ORF72 iPSC-derived motor neurons are rescued due to, surprisingly, the decreased levels of dipeptide repeat (DPR) proteins and expanded G4C2 repeats-containing RNA. The treatment of fully differentiated C9ORF72 neurons with EXOC2 antisense oligonucleotides also decreases expanded G4C2 repeats-containing RNA and partially rescued disease phenotypes. These results indicate that EXOC2 directly or indirectly regulates the level of G4C2 repeats-containing RNA, making it a potential therapeutic target in C9ORF72-ALS/FTD.
Generation and characterization of monoclonal antibodies against pathologically phosphorylated TDP-43.
Inclusions containing TAR DNA binding protein 43 (TDP-43) are a pathological hallmark of frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS). One of the disease-specific features of TDP-43 inclusions is the aberrant phosphorylation of TDP-43 at serines 409/410 (pS409/410). Here, we developed rabbit monoclonal antibodies (mAbs) that specifically detect pS409/410-TDP-43 in multiple model systems and FTD/ALS patient samples. Specifically, we identified three mAbs (26H10, 2E9 and 23A1) from spleen B cell clones that exhibit high specificity and sensitivity to pS409/410-TDP-43 peptides in an ELISA assay. Biochemical analyses revealed that pS409/410 of recombinant TDP-43 and of exogenous 25 kDa TDP-43 C-terminal fragments in cultured HEK293T cells are detected by all three mAbs. Moreover, the mAbs detect pS409/410-positive TDP-43 inclusions in the brains of FTD/ALS patients and mouse models of TDP-43 proteinopathy by immunohistochemistry. Our findings indicate that these mAbs are a valuable resource for investigating TDP-43 pathology both in vitro and in vivo.
Publicações recentes
[An autopsy case report of a patient with frontotemporal dementia with motor neuron disease in totally locked-in state showing hyperosmolar hyperosmotic state].
Increased Neurofilament Light Chain and YKL-40 CSF Levels in One Japanese IBMPFD Patient With VCP R155C Mutation: A Clinical Case Report With CSF Biomarker Analyses.
Detection of reduced dopamine transporter availability by (123) I-N-omega-fluoropropyl-2-beta-carbomethoxy-3-beta (4-iodophenyl) nortropane single-photon emission computed tomography in a patient of frontotemporal dementia with motor neuron disease.
Neuroimaging in aging and neurologic diseases.
📚 EuropePMC18 artigos no totalmostrando 142
Matrin-3 forms spherical and wormlike assemblies that are modulated by RNA binding and ALS/FTD-associated mutations.
Molecular cellA high-fidelity CRISPR-Cas13 system improves abnormalities associated with C9ORF72-linked ALS/FTD.
Nature communicationsInterference of nuclear speckles: A nexus of RNA foci, dipeptide repeats, and mis-splicing in C9ORF72 ALS/FTD.
NeuronPoly-GR repeats associated with ALS/FTD gene C9ORF72 impair translation elongation and induce a ribotoxic stress response in neurons.
Science signalingThe exocyst subunit EXOC2 regulates the toxicity of expanded GGGGCC repeats in C9ORF72-ALS/FTD.
Cell reportsCrystal structure of a tetrameric RNA G-quadruplex formed by hexanucleotide repeat expansions of C9orf72 in ALS/FTD.
Nucleic acids researchC9orf72-Associated Dipeptide Repeat Expansions Perturb ER-Golgi Vesicular Trafficking, Inducing Golgi Fragmentation and ER Stress, in ALS/FTD.
Molecular neurobiologyLINC complex alterations are a key feature of sporadic and familial ALS/FTD.
Acta neuropathologica communicationsGeneration and characterization of monoclonal antibodies against pathologically phosphorylated TDP-43.
PloS oneStimulating VAPB-PTPIP51 ER-mitochondria tethering corrects FTD/ALS mutant TDP43 linked Ca2+ and synaptic defects.
Acta neuropathologica communicationsRepeated mild traumatic brain injury triggers pathology in asymptomatic C9ORF72 transgenic mice.
Brain : a journal of neurologyYoung Onset Alzheimer's Disease Associated with C9ORF72 Hexanucleotide Expansion: Further Evidence for a Still Unsolved Association.
GenesAntisense, but not sense, repeat expanded RNAs activate PKR/eIF2α-dependent ISR in C9ORF72 FTD/ALS.
eLifeInflammasome-Mediated Neuronal-Microglial Crosstalk: a Therapeutic Substrate for the Familial C9orf72 Variant of Frontotemporal Dementia/Amyotrophic Lateral Sclerosis.
Molecular neurobiologyGasdermin-E mediates mitochondrial damage in axons and neurodegeneration.
NeuronReversing lysosome-ribosome circuit dysregulation mitigates C9FTD/ALS neurodegeneration and behaviors.
Human molecular geneticsTwo FTD-ALS genes converge on the endosomal pathway to induce TDP-43 pathology and degeneration.
Science (New York, N.Y.)[An autopsy case report of a patient with frontotemporal dementia with motor neuron disease in totally locked-in state showing hyperosmolar hyperosmotic state].
Rinsho shinkeigaku = Clinical neurologyModulation of synaptic plasticity, motor unit physiology, and TDP-43 pathology by CHCHD10.
Acta neuropathologica communicationsSchizotypal traits across the amyotrophic lateral sclerosis-frontotemporal dementia spectrum: pathomechanistic insights.
Journal of neurologyTDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A.
NatureDifferential fates of introns in gene expression due to global alternative splicing.
Human geneticsTrichostatin A Relieves Growth Suppression and Restores Histone Acetylation at Specific Sites in a FUS ALS/FTD Yeast Model.
BiochemistryA C. elegans model of C9orf72-associated ALS/FTD uncovers a conserved role for eIF2D in RAN translation.
Nature communicationsThe nuclear ubiquitin ligase adaptor SPOP is a conserved regulator of C9orf72 dipeptide toxicity.
Proceedings of the National Academy of Sciences of the United States of AmericaTDP-43 stabilizes G3BP1 mRNA: relevance to amyotrophic lateral sclerosis/frontotemporal dementia.
Brain : a journal of neurologyImpaired 26S Proteasome Assembly Precedes Neuronal Loss in Mutant UBQLN2 Rats.
International journal of molecular sciencesCharacterization of C9orf72 haplotypes to evaluate the effects of normal and pathological variations on its expression and splicing.
PLoS geneticsSerpin neuropathology in the P497S UBQLN2 mouse model of ALS/FTD.
Brain pathology (Zurich, Switzerland)An autopsy case of pure nigropathy with TUBA4A nonsense mutation.
Neuropathology and applied neurobiologySynaptic dysfunction in amyotrophic lateral sclerosis/frontotemporal dementia: Therapeutic strategies and novel biomarkers.
Journal of neuroscience researchAltered network properties in C9ORF72 repeat expansion cortical neurons are due to synaptic dysfunction.
Molecular neurodegenerationC9orf72 ALS-FTD: recent evidence for dysregulation of the autophagy-lysosome pathway at multiple levels.
AutophagyInduction of autophagy mitigates TDP-43 pathology and translational repression of neurofilament mRNAs in mouse models of ALS/FTD.
Molecular neurodegenerationDetection of the SQSTM1 Mutation in a Patient with Early-Onset Hippocampal Amnestic Syndrome.
Journal of Alzheimer's disease : JADA novel temporal-predominant neuro-astroglial tauopathy associated with TMEM106B gene polymorphism in FTLD/ALS-TDP.
Brain pathology (Zurich, Switzerland)Global proteomics of Ubqln2-based murine models of ALS.
The Journal of biological chemistryCellular and physiological functions of C9ORF72 and implications for ALS/FTD.
Journal of neurochemistryRepeat RNA expansion disorders of the nervous system: post-transcriptional mechanisms and therapeutic strategies.
Critical reviews in biochemistry and molecular biologyALS/FTLD-Linked Mutations in FUS Glycine Residues Cause Accelerated Gelation and Reduced Interactions with Wild-Type FUS.
Molecular cellSQSTM1 variant in disorders of the frontotemporal dementia-amyotrophic lateral sclerosis spectrum: identification of a novel heterozygous variant and a review of the literature.
Journal of neurologyWidespread intron retention impairs protein homeostasis in C9orf72 ALS brains.
Genome researchOverexpression of UBQLN1 reduces neuropathology in the P497S UBQLN2 mouse model of ALS/FTD.
Acta neuropathologica communicationsC9orf72 Gene Expression in Frontotemporal Dementia and Amyotrophic Lateral Sclerosis.
Bulletin of experimental biology and medicineQuality-control mechanisms targeting translationally stalled and C-terminally extended poly(GR) associated with ALS/FTD.
Proceedings of the National Academy of Sciences of the United States of AmericaC9orf72 loss-of-function: a trivial, stand-alone or additive mechanism in C9 ALS/FTD?
Acta neuropathologicaIncreased Neurofilament Light Chain and YKL-40 CSF Levels in One Japanese IBMPFD Patient With VCP R155C Mutation: A Clinical Case Report With CSF Biomarker Analyses.
Frontiers in neurologyStructure of the C9orf72 ARF GAP complex that is haploinsufficient in ALS and FTD.
NatureAntibody against TDP-43 phosphorylated at serine 375 suggests conformational differences of TDP-43 aggregates among FTLD-TDP subtypes.
Acta neuropathologicaThe carboxyl termini of RAN translated GGGGCC nucleotide repeat expansions modulate toxicity in models of ALS/FTD.
Acta neuropathologica communicationsThe role of hnRNPs in frontotemporal dementia and amyotrophic lateral sclerosis.
Acta neuropathologicaG4C2 Repeat RNA Initiates a POM121-Mediated Reduction in Specific Nucleoporins in C9orf72 ALS/FTD.
NeuronModeling UBQLN2-mediated neurodegenerative disease in mice: Shared and divergent properties of wild type and mutant UBQLN2 in phase separation, subcellular localization, altered proteostasis pathways, and selective cytotoxicity.
Neurobiology of diseaseC9orf72 arginine-rich dipeptide repeats inhibit UPF1-mediated RNA decay via translational repression.
Nature communicationsDetection of reduced dopamine transporter availability by 123 I-N-omega-fluoropropyl-2-beta-carbomethoxy-3-beta (4-iodophenyl) nortropane single-photon emission computed tomography in a patient of frontotemporal dementia with motor neuron disease.
Psychogeriatrics : the official journal of the Japanese Psychogeriatric SocietyLoss of CHCHD2 and CHCHD10 activates OMA1 peptidase to disrupt mitochondrial cristae phenocopying patient mutations.
Human molecular geneticsReduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72.
Nature neuroscienceCRISPR deletion of the C9ORF72 promoter in ALS/FTD patient motor neurons abolishes production of dipeptide repeat proteins and rescues neurodegeneration.
Acta neuropathologicaDevelopment of disease-modifying drugs for frontotemporal dementia spectrum disorders.
Nature reviews. NeurologyTraffic jam at the nuclear pore: All roads lead to nucleocytoplasmic transport defects in ALS/FTD.
Neurobiology of diseaseDipeptide repeat proteins inhibit homology-directed DNA double strand break repair in C9ORF72 ALS/FTD.
Molecular neurodegenerationErbB4 Mutation that Decreased NRG1-ErbB4 Signaling Involved in the Pathogenesis of Amyotrophic Lateral Sclerosis/Frontotemporal Dementia.
Journal of Alzheimer's disease : JADNucleocytoplasmic Proteomic Analysis Uncovers eRF1 and Nonsense-Mediated Decay as Modifiers of ALS/FTD C9orf72 Toxicity.
NeuronCognitive and behavioral status in Japanese ALS patients: a multicenter study.
Journal of neurologyReduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders.
The EMBO journalCSF angiogenin levels in amyotrophic lateral Sclerosis-Frontotemporal dementia spectrum.
Amyotrophic lateral sclerosis & frontotemporal degenerationMotor function decline correlates with behavioral impairment in C9orf72 mutation carriers.
NeurologyAmyotrophic lateral sclerosis-linked UBQLN2 mutants inhibit endoplasmic reticulum to Golgi transport, leading to Golgi fragmentation and ER stress.
Cellular and molecular life sciences : CMLSThree-Dimensional Structure of RNA Monomeric G-Quadruplex Containing ALS and FTD Related G4C2 Repeat and Its Binding with TMPyP4 Probed by Homology Modeling based on Experimental Constraints and Molecular Dynamics Simulations.
ACS chemical neuroscienceTranscription elongation factor AFF2/FMR2 regulates expression of expanded GGGGCC repeat-containing C9ORF72 allele in ALS/FTD.
Nature communicationsNeuroimaging in aging and neurologic diseases.
Handbook of clinical neurologyRNA toxicity in non-coding repeat expansion disorders.
The EMBO journalTargeted DNA methylation of neurodegenerative disease genes via homology directed repair.
Nucleic acids researchStress granule mediated protein aggregation and underlying gene defects in the FTD-ALS spectrum.
Neurobiology of diseasePhenotypic Suppression of ALS/FTD-Associated Neurodegeneration Highlights Mechanisms of Dysfunction.
The Journal of neuroscience : the official journal of the Society for NeuroscienceAntisense therapies for movement disorders.
Movement disorders : official journal of the Movement Disorder SocietySmall-Molecule Modulation of TDP-43 Recruitment to Stress Granules Prevents Persistent TDP-43 Accumulation in ALS/FTD.
NeuronC9ORF72-ALS/FTD-associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo.
Nature neuroscienceL3MBTL1 regulates ALS/FTD-associated proteotoxicity and quality control.
Nature neuroscienceC9orf72 and triplet repeat disorder RNAs: G-quadruplex formation, binding to PRC2 and implications for disease mechanisms.
RNA (New York, N.Y.)Loss of Nuclear TDP-43 Is Associated with Decondensation of LINE Retrotransposons.
Cell reportsIncidence of frontotemporal lobar degeneration in Italy: The Salento-Brescia Registry study.
NeurologyADAR2 mislocalization and widespread RNA editing aberrations in C9orf72-mediated ALS/FTD.
Acta neuropathologicaChronic optogenetic induction of stress granules is cytotoxic and reveals the evolution of ALS-FTD pathology.
eLifeSpeech and language intervention for language impairment in patients in the FTD-ALS spectrum.
Hellenic journal of nuclear medicineMitochondrial defect in muscle precedes neuromuscular junction degeneration and motor neuron death in CHCHD10S59L/+ mouse.
Acta neuropathologicaMolecular Mechanisms of Neurodegeneration Related to C9orf72 Hexanucleotide Repeat Expansion.
Behavioural neurologyValidation of the revised classification of cognitive and behavioural impairment in ALS.
Journal of neurology, neurosurgery, and psychiatrySMCR8 negatively regulates AKT and MTORC1 signaling to modulate lysosome biogenesis and tissue homeostasis.
AutophagyThe ALS-FTD-linked gene product, C9orf72, regulates neuronal morphogenesis via autophagy.
AutophagyAntisense RNA foci are associated with nucleoli and TDP-43 mislocalization in C9orf72-ALS/FTD: a quantitative study.
Acta neuropathologicaScreening for cognitive and behavioral change in amyotrophic lateral sclerosis/motor neuron disease: a systematic review of validated screening methods.
Amyotrophic lateral sclerosis & frontotemporal degenerationA Chemical Screen Identifies Compounds Limiting the Toxicity of C9ORF72 Dipeptide Repeats.
Cell chemical biologyThe Hairpin Form of r(G4C2)exp in c9ALS/FTD Is Repeat-Associated Non-ATG Translated and a Target for Bioactive Small Molecules.
Cell chemical biologyMeasurement of spinal cord atrophy using phase sensitive inversion recovery (PSIR) imaging in motor neuron disease.
PloS oneProgranulin reduces insoluble TDP-43 levels, slows down axonal degeneration and prolongs survival in mutant TDP-43 mice.
Molecular neurodegenerationA feedback loop between dipeptide-repeat protein, TDP-43 and karyopherin-α mediates C9orf72-related neurodegeneration.
Brain : a journal of neurologyRepeat-associated non-ATG (RAN) translation.
The Journal of biological chemistryLoss of MICOS complex integrity and mitochondrial damage, but not TDP-43 mitochondrial localisation, are likely associated with severity of CHCHD10-related diseases.
Neurobiology of diseaseTDP-43 as a potential biomarker for amyotrophic lateral sclerosis: a systematic review and meta-analysis.
BMC neurologyLost in Translation: Evidence for Protein Synthesis Deficits in ALS/FTD and Related Neurodegenerative Diseases.
Advances in neurobiologyA C9orf72 ALS/FTD Ortholog Acts in Endolysosomal Degradation and Lysosomal Homeostasis.
Current biology : CBAll in the Family: Repeats and ALS/FTD.
Trends in neurosciencesStress Granule Assembly Disrupts Nucleocytoplasmic Transport.
CellIntron retention induced by microsatellite expansions as a disease biomarker.
Proceedings of the National Academy of Sciences of the United States of AmericaExploring the genetics and non-cell autonomous mechanisms underlying ALS/FTLD.
Cell death and differentiationTraumatic injury induces stress granule formation and enhances motor dysfunctions in ALS/FTD models.
Human molecular geneticsSelf-assembly of FUS through its low-complexity domain contributes to neurodegeneration.
Human molecular geneticsHaploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons.
Nature medicineOligogenic genetic variation of neurodegenerative disease genes in 980 postmortem human brains.
Journal of neurology, neurosurgery, and psychiatryIs amyotrophic lateral sclerosis/frontotemporal dementia an autophagy disease?
Molecular neurodegenerationLipid Metabolism and Survival Across the Frontotemporal Dementia-Amyotrophic Lateral Sclerosis Spectrum: Relationships to Eating Behavior and Cognition.
Journal of Alzheimer's disease : JADA proteomic network approach across the ALS-FTD disease spectrum resolves clinical phenotypes and genetic vulnerability in human brain.
EMBO molecular medicineMutation-dependent aggregation and toxicity in a Drosophila model for UBQLN2-associated ALS.
Human molecular geneticsRNA binding proteins and the pathological cascade in ALS/FTD neurodegeneration.
Science translational medicineG-quadruplex-binding small molecules ameliorate C9orf72 FTD/ALS pathology in vitro and in vivo.
EMBO molecular medicineOPTN p.Met468Arg and ATXN2 intermediate length polyQ extension in families with C9orf72 mediated amyotrophic lateral sclerosis and frontotemporal dementia.
American journal of medical genetics. Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric GeneticsNEK1 genetic variability in a Belgian cohort of ALS and ALS-FTD patients.
Neurobiology of agingAmyotrophic lateral sclerosis modifies progenitor neural proliferation in adult classic neurogenic brain niches.
BMC neurologyPathogenic mutation in the ALS/FTD gene, CCNF, causes elevated Lys48-linked ubiquitylation and defective autophagy.
Cellular and molecular life sciences : CMLSImmunohistochemical detection of C9orf72 protein in frontotemporal lobar degeneration and motor neurone disease: patterns of immunostaining and an evaluation of commercial antibodies.
Amyotrophic lateral sclerosis & frontotemporal degenerationCortical influences drive amyotrophic lateral sclerosis.
Journal of neurology, neurosurgery, and psychiatrySRSF1-dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits.
Nature communicationsMotoneuron Disease: Clinical.
Advances in neurobiologyEvidence that C9ORF72 Dipeptide Repeat Proteins Associate with U2 snRNP to Cause Mis-splicing in ALS/FTD Patients.
Cell reportsLoss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity.
Nature communicationsExpression of ALS/FTD-linked mutant CCNF in zebrafish leads to increased cell death in the spinal cord and an aberrant motor phenotype.
Human molecular genetics2D and 3D FISH of expanded repeat RNAs in human lymphoblasts.
Methods (San Diego, Calif.)New developments in RAN translation: insights from multiple diseases.
Current opinion in genetics & developmentGenetic compendium of 1511 human brains available through the UK Medical Research Council Brain Banks Network Resource.
Genome researchFrontotemporal Dementia with Motor Neuron Disease in a Patient with Antiphospholipid Syndrome: A Case Report.
Dementia and neurocognitive disordersIncreased prevalence of autoimmune disease within C9 and FTD/MND cohorts: Completing the picture.
Neurology(R) neuroimmunology & neuroinflammationTBK1 is associated with ALS and ALS-FTD in Sardinian patients.
Neurobiology of agingSyntactic comprehension deficits across the FTD-ALS continuum.
Neurobiology of agingLoss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death.
The EMBO journalPoor Gait Performance and Prediction of Dementia: Results From a Meta-Analysis.
Journal of the American Medical Directors AssociationAn amyloid-like cascade hypothesis for C9orf72 ALS/FTD.
Current opinion in neurobiologyC9orf72 expansion presenting as an eating disorder.
Journal of clinical neuroscience : official journal of the Neurosurgical Society of AustralasiaNovel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72.
Acta neuropathologicaQuantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers.
Acta neuropathologicaCognitive and Behavioral Symptoms in ALSFTD: Detection, Differentiation, and Progression.
Journal of geriatric psychiatry and neurologyDefining the genetic connection linking amyotrophic lateral sclerosis (ALS) with frontotemporal dementia (FTD).
Trends in genetics : TIGAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Demência frontotemporal com doença do neurônio motor.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Demência frontotemporal com doença do neurônio motor
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Matrin-3 forms spherical and wormlike assemblies that are modulated by RNA binding and ALS/FTD-associated mutations.
- A high-fidelity CRISPR-Cas13 system improves abnormalities associated with C9ORF72-linked ALS/FTD.
- Poly-GR repeats associated with ALS/FTD gene C9ORF72 impair translation elongation and induce a ribotoxic stress response in neurons.
- The exocyst subunit EXOC2 regulates the toxicity of expanded GGGGCC repeats in C9ORF72-ALS/FTD.
- Generation and characterization of monoclonal antibodies against pathologically phosphorylated TDP-43.
- [An autopsy case report of a patient with frontotemporal dementia with motor neuron disease in totally locked-in state showing hyperosmolar hyperosmotic state].
- Increased Neurofilament Light Chain and YKL-40 CSF Levels in One Japanese IBMPFD Patient With VCP R155C Mutation: A Clinical Case Report With CSF Biomarker Analyses.
- Detection of reduced dopamine transporter availability by (123) I-N-omega-fluoropropyl-2-beta-carbomethoxy-3-beta (4-iodophenyl) nortropane single-photon emission computed tomography in a patient of frontotemporal dementia with motor neuron disease.
- Neuroimaging in aging and neurologic diseases.
- Frontotemporal Dementia.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:275872(Orphanet)
- MONDO:0017161(MONDO)
- GARD:17273(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q3705268(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Demência frontotemporal com doença do neurônio motor
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov