A deficiência da GM3 sintase é caracterizada por crises epilépticas recorrentes (epilepsia) e problemas no desenvolvimento do cérebro. Nas primeiras semanas de vida, os bebês afetados ficam irritados e desenvolvem dificuldades para se alimentar e vômitos, o que os impede de crescer e ganhar peso no ritmo normal. As crises epilépticas começam no primeiro ano de vida e pioram com o tempo. Podem ocorrer vários tipos de crises, incluindo as crises tônico-clônicas generalizadas (também conhecidas como crises de grande mal), que causam rigidez muscular, convulsões e perda de consciência. Algumas crianças afetadas também apresentam episódios prolongados de atividade epiléptica, chamados de estado de mal epiléptico não convulsivo. As crises relacionadas à deficiência da GM3 sintase tendem a ser resistentes (refratárias) ao tratamento com medicamentos antiepilépticos.
Introdução
O que você precisa saber de cara
A deficiência da GM3 sintase é caracterizada por crises epilépticas recorrentes (epilepsia) e problemas no desenvolvimento do cérebro. Nas primeiras semanas de vida, os bebês afetados ficam irritados e desenvolvem dificuldades para se alimentar e vômitos, o que os impede de crescer e ganhar peso no ritmo normal. As crises epilépticas começam no primeiro ano de vida e pioram com o tempo. Podem ocorrer vários tipos de crises, incluindo as crises tônico-clônicas generalizadas (também conhecidas como crises de grande mal), que causam rigidez muscular, convulsões e perda de consciência. Algumas crianças afetadas também apresentam episódios prolongados de atividade epiléptica, chamados de estado de mal epiléptico não convulsivo. As crises relacionadas à deficiência da GM3 sintase tendem a ser resistentes (refratárias) ao tratamento com medicamentos antiepilépticos.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 7 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 26 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição.
Curadoria gene-doença
fontes oficiaisTransfers the sialyl group (N-acetyl-alpha-neuraminyl or NeuAc) from CMP-NeuAc to the non-reducing terminal galactose (Gal) of glycosphingolipids forming gangliosides (important molecules involved in the regulation of multiple cellular processes, including cell proliferation and differentiation, apoptosis, embryogenesis, development, and oncogenesis) (PubMed:16934889, PubMed:9822625). Mainly involved in the biosynthesis of ganglioside GM3 but can also use different glycolipids as substrate accep
Golgi apparatus membrane
Salt and pepper developmental regression syndrome
A rare autosomal recessive disorder characterized by infantile onset of severe, recurrent and refractory seizures, failure to thrive, psychomotor delay, developmental stagnation, and cortical blindness. Deafness is observed in some patients. Affected individuals have patches of skin hypo- or hyperpigmentation on the trunk, face, and extremities.
Variantes genéticas (ClinVar)
75 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de sal e pimenta
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Two Cases of CLIPPERS-like Syndrome Sharing a Hypomorphic UNC13D Variant.
Chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids (CLIPPERS) syndrome is a central nervous system (CNS) inflammatory disease characterized by contrast-enhanced MRI findings of salt-and-pepper-like lesions predominantly affecting the brainstem and cerebellum. We report two patients with CLIPPERS-like brain MRI findings who carried the same missense UNC13D variant in one allele along with deleterious variants in the opposite allele. Patient 1, a 23-year-old female, presented at 15 years of age with neurological symptoms and an MRI showing spontaneously resolving, contrast-enhancing lesions in the cerebellum. At age 16, the patient experienced an episode with systemic manifestations followed by recurrent CNS lesions that responded to steroid therapy. At age 22, the patient developed punctate to nodular contrast-enhancing lesions in the brainstem, cerebellum, and cerebrum, findings consistent with CLIPPERS. Patient 2, an 18-year-old female, presented at age 11 with ataxia and dysarthria, and an MRI showing multiple contrast-enhancing lesions in the cerebellum and brainstem, consistent with CLIPPERS MRI findings. Cerebellar biopsy revealed perivascular CD4+ T-lymphocyte infiltration, and the patient responded to steroid therapy, leading to an initial diagnosis of CLIPPERS. These patients were suspected of having inborn errors of immunity and were identified to have compound heterozygous UNC13D variants along with downregulated Munc13-4 protein. Both patients underwent allogeneic hematopoietic cell transplantation, with patient 1 remaining neurologically stable for two years post-transplantation, while patient 2 experienced a post-transplant relapse requiring steroid therapy. These cases highlight that biallelic variants in the UNC13D gene may cause CNS-predominant inflammation that mimics CLIPPERS. [Image: see text] The online version contains supplementary material available at 10.1007/s10875-026-01988-1.
Severe Manifestation of Hyperparathyroidism With Sagliker Syndrome: A Case Report.
Sagliker syndrome represents a rare and severe manifestation of secondary renal osteodystrophy in patients with end-stage chronic kidney disease. We present the case of a 26-year-old woman on peritoneal dialysis with a one-year history of progressive craniofacial deformity involving both the maxilla and mandible. Laboratory tests demonstrated markedly elevated parathyroid hormone levels (1,936 pg/mL). Cranial CT revealed a diffuse "salt and pepper" pattern, serpiginous lytic trabecular changes, and focal ground-glass areas in the calvarium. Correlation of severe hyperparathyroidism with these characteristic imaging features confirmed Sagliker syndrome. Early recognition is essential to prevent irreversible deformities and long-term morbidity. The patient was managed through a multidisciplinary approach, including medical optimization of secondary hyperparathyroidism, followed by total parathyroidectomy due to persistent biochemical abnormalities and progressive craniofacial deformity.
How the diffuse neuroendocrine system shapes health, homeostasis, and cancer.
The diffuse neuroendocrine system (DNES) consists of dispersed neuroendocrine (NE) cells that bridge nervous, immune, and endocrine pathways across organs. Evolutionarily, DNES traces to primitive metazoans where single cells combined neural and immune roles, later diversifying into specialized vertebrate NE cells. Hallmark traits include dense-core granules, amine metabolism, "salt-and-pepper" chromatin, and regulation by ASCL1, NEUROG3, and INSM1. Remarkable plasticity allows immune and epithelial cells to acquire NE features under stress, while carcinomas exploit this program to form aggressive neuroendocrine tumors (NETs) and resist therapy. Canonical neuroimmune circuits, the Vagus-driven inflammatory reflex and hypothalamic-pituitary-adrenal stress axis, illustrate DNES coordination of systemic responses. Clinically, DNES-derived neoplasms span multiple organs, produce diverse hormonal syndromes, and are managed with somatostatin analogues, epigenetic drugs, and emerging immunotherapies. Recognizing DNES as a diffuse, integrative regulatory network clarifies mechanisms of chronic inflammation and cancer evolution and offers novel therapeutic entry points for disorders ranging from asthma to pancreatic neuroendocrine carcinomas.
Advances in imaging techniques for Sjogren's disease.
Imaging of salivary glands (SG), particularly Salivary Glands Ultrasonography (SGUS) is increasingly used in patients with suspected Sjogren's disease (SD). SGUS is the first-line imaging modality. Numerous studies have highlighted this non-invasive, non-irradiating, and low-cost imaging modality. The OMERACT group has established a classification of SG structural damage based on B-mode findings, ranging from grade 0 (normal) to 3 (severe structural damage). SGUS abnormalities (≥ grade 2) have been reported in approximately 63 % of patients with SD. More recently, Hocevar et al. described a Doppler-based classification assessing SG parenchymal vascularization, graded from 0 (normal) to 3 (Doppler signals occupying the entire glandular surface) and could be used as a marker of disease activity and as a biomarker of response to therapy. Moreover, SGUS can be useful for looking for complications such as lymphoma. New ultrasound techniques are currently being developed, including elastography for assessing tissue stiffness, analysis of microvascularization using contrast-enhanced ultrasound with microbubbles, and analysis of minor salivary glands using the ultra-high frequency probe. The combination of several US modalities enhances both sensitivity and specificity of the technique, allowing for the development of a comprehensive multimodal imaging approach. Other imaging techniques can be performed for SD, such as MRI of the parotid glands, allowing analysis of the glandular parenchyma ("salt and pepper" appearance), and certain sequences (DWI-MR) should be performed when lymphoma or other tumors are suspected. 18-FDG PET-CT may be useful to detect systemic manifestations or complications in SD and new PET tracers are currently being developed.
Rubella Retinopathy.
Rubella retinopathy is an ocular manifestation of congenital rubella syndrome characterized by diffuse mottling of the retinal pigment epithelium classically described as a "salt-and-pepper" fundus.
Publicações recentes
A novel case of Heimler syndrome in a young child with compound heterozygous PEX26 mutations: clinical and genetic insights with literature review.
Ophthalmic manifestations of Danon disease: A systematic review.
Ampullary Amphicrine Carcinoma: A Rare Case Report With Review of Literature.
Two Cases of CLIPPERS-like Syndrome Sharing a Hypomorphic UNC13D Variant.
Establishment of patient-derived xenografts for neuroendocrine tumors in the avian embryo model.
📚 EuropePMC2 artigos no totalmostrando 35
Two Cases of CLIPPERS-like Syndrome Sharing a Hypomorphic UNC13D Variant.
Journal of clinical immunologySevere Manifestation of Hyperparathyroidism With Sagliker Syndrome: A Case Report.
CureusHow the diffuse neuroendocrine system shapes health, homeostasis, and cancer.
Frontiers in neuroendocrinologyAdvances in imaging techniques for Sjogren's disease.
Best practice & research. Clinical rheumatologyWell-Differentiated Pancreatic Neuroendocrine Tumor: Does Morphologic Variant Matter?
Archives of pathology & laboratory medicineUnmasking Villaret's syndrome: a diagnostic challenge of glomus jugulare mimicking mastoiditis.
Annals of medicine and surgery (2012)Retinopathy associated with MELAS syndrome. A case report.
Archivos de la Sociedad Espanola de OftalmologiaSalt and Pepper Parotid Changes in Sjögren's Syndrome.
European journal of rheumatologyAn unusual duo: Immunodeficiency disorder and scleroderma.
Indian journal of sexually transmitted diseases and AIDSChronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids (CLIPPERS): contemporary advances and current controversies.
Journal of neurologyFacial dysmorphism in congenital rubella syndrome.
Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and StrabismusFundus imaging features of congenital rubella retinopathy.
Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle OphthalmologiePigmentary retinopathy and nodular granuloma associated with acute retinal necrosis from varicella zoster virus and human herpes virus type 6: Case report.
MedicineSalt-and-Pepper Dyspigmentation with Groove Sign in Erasmus Syndrome: A Double Jeopardy.
Indian dermatology online journalOphthalmoplegia, pigmentary retinopathy, and abnormal cardiac conduction: A rare case of Kearns-Sayre syndrome.
eNeurologicalSciWhole Exome Sequencing Reveals a Novel Homozygous Variant in the Ganglioside Biosynthetic Enzyme, ST3GAL5 Gene in a Saudi Family Causing Salt and Pepper Syndrome.
GenesA rare case report of primary Sjögren's syndrome with clinical characteristics similar to those of CLIPPERS.
BMC neurologyClinical and histopathological principles for the diagnosis of a recurrent paraganglioma of the jugular foramen initially diagnosed as a middle ear adenoma: illustrative case.
Journal of neurosurgery. Case lessonsErasmus Syndrome: A Case Report and Literature Review.
The American journal of case reportsSeropositive Neuromyelitis Optica in a Case of Undiagnosed Ankylosing Spondylitis: A Neuro-Rheumatological Conundrum.
Qatar medical journalAtypical painful stroke presentations: A review.
Acta neurologica ScandinavicaChronic Lymphocytic Inflammation With Pontine Perivascular Enhancement Responsive to Steroids: An Acute Presentation.
CureusNewborn Glaucoma: A Neglected Manifestation of Congenital Rubella Syndrome.
Ophthalmology. GlaucomaPrimary thoracic gastrinoma causing Zollinger-Ellison syndrome.
Indian journal of thoracic and cardiovascular surgeryA novel frameshift pathogenic variant in ST3GAL5 causing salt and pepper developmental regression syndrome (SPDRS): A case report.
Human genome variationRetinitis Pigmentosa and Polydactyly in a Patient with a Heterozygous Mutation on the BBS1 Gene.
International medical case reports journalCoexistence of bilateral macular edema and pale optic disc in the patient with Cohen syndrome.
Open medicine (Warsaw, Poland)Prediction of probability of rubella based on eye outcomes (PORBEO Nomogram)-a cross-sectional sentinel surveillance of 1134 infants.
Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle OphthalmologieRadiological imaging features of the salivary glands in xerostomia induced by an immune checkpoint inhibitor.
Oral radiologyA Prospective OCT Study of Rubella Retinopathy.
Ophthalmology. RetinaMitochondrial Disorder: Kearns-Sayre Syndrome.
Advances in experimental medicine and biologyEF-hand domain containing 2 (Efhc2) is crucial for distal segmentation of pronephros in zebrafish.
Cell & bioscienceClinical utility of hypo- and hyperpigmentation of skin in diffuse cutaneous systemic sclerosis.
International journal of rheumatic diseasesDisruption of the Photoreceptor Inner Segment-Outer Segment Junction in a 6-Year-Old Girl with Joubert Syndrome.
Neuro-ophthalmology (Aeolus Press)Effectiveness of imaging modalities for screening IgG4-related dacryoadenitis and sialadenitis (Mikulicz's disease) and for differentiating it from Sjögren's syndrome (SS), with an emphasis on sonography.
Arthritis research & therapyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de sal e pimenta.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de sal e pimenta
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Two Cases of CLIPPERS-like Syndrome Sharing a Hypomorphic UNC13D Variant.
- Severe Manifestation of Hyperparathyroidism With Sagliker Syndrome: A Case Report.
- How the diffuse neuroendocrine system shapes health, homeostasis, and cancer.
- Advances in imaging techniques for Sjogren's disease.
- Rubella Retinopathy.
- A novel case of Heimler syndrome in a young child with compound heterozygous PEX26 mutations: clinical and genetic insights with literature review.
- Ophthalmic manifestations of Danon disease: A systematic review.
- Ampullary Amphicrine Carcinoma: A Rare Case Report With Review of Literature.
- Establishment of patient-derived xenografts for neuroendocrine tumors in the avian embryo model.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:370938(Orphanet)
- OMIM OMIM:609056(OMIM)
- MONDO:0018274(MONDO)
- GARD:12059(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q21505494(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
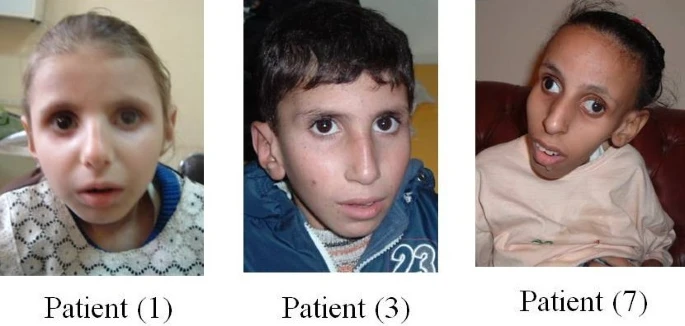
Síndrome de sal e pimenta
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata