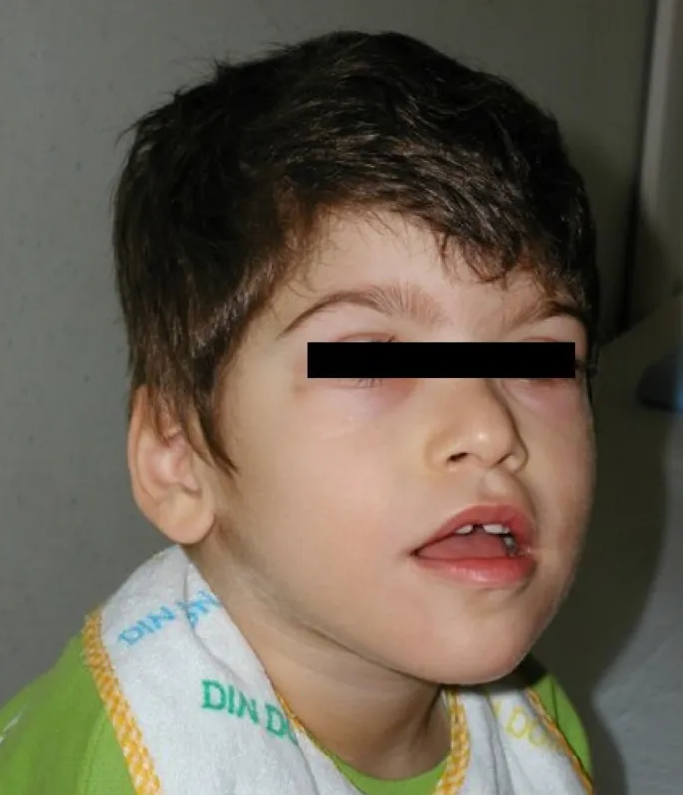
A síndrome de Feingold tipo 2 (FS2) é uma síndrome de malformação hereditária rara caracterizada por anormalidades esqueléticas e deficiência intelectual leve semelhantes às observadas na síndrome de Feingold tipo 1 (FS1), mas que não apresenta manifestações de atresia gastrointestinal e fissuras palpebrais curtas.
Introdução
O que você precisa saber de cara
A síndrome de Feingold tipo 2 (FS2) é uma síndrome de malformação hereditária rara caracterizada por anormalidades esqueléticas e deficiência intelectual leve semelhantes às observadas na síndrome de Feingold tipo 1 (FS1), mas que não apresenta manifestações de atresia gastrointestinal e fissuras palpebrais curtas.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 3 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 23 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisMembrane
Feingold syndrome 2
A syndrome characterized by microcephaly, short stature, and digital abnormalities including brachydactyly, brachymesophalangy of the second and fifth fingers, hypoplastic thumbs of variable severity, and cutaneous syndactyly of the toes.
Variantes genéticas (ClinVar)
86 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 124 variantes classificadas pelo ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de Feingold, tipo 2
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
First Patient Diagnosed as Feingold Syndrome Type 2 with Alport Syndrome and Review of the Current Literature.
Feingold syndrome type 2 (FGLDS2) is an ultra-rare genetic disorder characterized by short stature, microcephaly, digital abnormalities, and intellectual disability. Until now, 22 patients have been reported in the literature. FGLDS2 is caused by a germline heterozygous deletion of 13q resulting in haploinsufficiency of the MIR17HG gene. In the present study, we evaluated clinical, radiological, and genetic analyses of a 10-year-old Turkish-origin girl with short stature, brachydactyly, intellectual disability, hematuria, and proteinuria. In the array-CGH analysis, a 15.7-Mb deletion, arr[hg19] 13q22q31.3(78,241,132_93,967,288)×1, was detected, and this alteration was evaluated to be pathogenic. The deletion of this region covering the MIR17HG gene is a potential cause of FGLDS2. Also, at her clinical exome sequencing study, a heterozygous c.2023G>A p.(Gly675Ser) variation was detected in the COL4A5 gene (NM_000495.4) that was likely pathogenic in up-to-date databases. As a result, we report on a patient who has FGLDS2 and Alport syndrome. This is the first report of a Turkish-origin FGLDS2 patient. Reporting new cases expands the range of phenotypes, plays a crucial role in understanding the FGLDS2 pathogenesis, and is important in terms of screening at-risk family members for giving appropriate genetic counseling and preimplantation genetic diagnosis opportunities.
Feingold syndrome type 2 in a patient from China.
Feingold syndrome type 2 (FGLDS2, MIM614326) is a genetic congenital malformation syndrome, caused by germline heterozygous deletion of MIR17HG on chromosome 13q31, which is extremely rare worldwide. To date, less than 25 patients have been described in the literature. Here, we report on a 3-year-old girl presented with hip dysplasia, polysyndactyly of the left thumb, brachymesophalangy of the fifth digit, microcephaly, intellectual disability, and growth delay. This is likely to be the first case of Feingold syndrome type 2 ever discovered among Chinese population. Through genetic testing and pedigree analysis, she was identified to have a de novo 4.8-Mb microdeletion at chromosome 13q31.3-q32.1, encompassing MIR17HG, GPC5, and GPC6. Additionally, we detected two common compound heterozygous variants (c.919-2A>G and c.147C>G) in SLC26A4 encoding pendrin protein, as well as a novel heterozygous variant c.985_988del in COMP encoding cartilage oligomeric matrix protein. This case report aims to analyze the microdeletion and the three types of variant detected in the patient, and to explore the association between the genotype and phenotype in patients with Feingold syndrome type 2, which may contribute to further understanding and future diagnosis of this disorder.
Distinct molecular pathways mediate Mycn and Myc-regulated miR-17-92 microRNA action in Feingold syndrome mouse models.
Feingold syndrome is a skeletal dysplasia caused by loss-of-function mutations of either MYCN (type 1) or MIR17HG that encodes miR-17-92 microRNAs (type 2). Since miR-17-92 expression is transcriptionally regulated by MYC transcription factors, it has been postulated that Feingold syndrome type 1 and 2 may be caused by a common molecular mechanism. Here we show that Mir17-92 deficiency upregulates TGF-β signaling, whereas Mycn-deficiency downregulates PI3K signaling in limb mesenchymal cells. Genetic or pharmacological inhibition of TGF-β signaling efficiently rescues the skeletal defects caused by Mir17-92 deficiency, suggesting that upregulation of TGF-β signaling is responsible for the skeletal defect of Feingold syndrome type 2. By contrast, the skeletal phenotype of Mycn-deficiency is partially rescued by Pten heterozygosity, but not by TGF-β inhibition. These results strongly suggest that despite the phenotypical similarity, distinct molecular mechanisms underlie the pathoetiology for Feingold syndrome type 1 and 2.
A case of Feingold type 2 syndrome associated with keratoconus refines keratoconus type 7 locus on chromosome 13q.
We report on a 58-year old woman with microcephaly, mild dysmorphic features, bilateral keratoconus, digital abnormalities, short stature and mild cognitive delay. Except for keratoconus, the phenotype was suggestive for Feingold syndrome type 2 (FGLDS2, MIM 614326), a rare autosomal dominant disorder described in six patients worldwide, due to the haploinsufficiency of MIR17HG, a micro RNA encoding gene. Karyotype showed a de novo deletion on chromosome 13q, further defined by array-Comparative Genomic Hybridization (a-CGH) to a 17.2-Mb region. The deletion included MIR17HG, as expected by the FGLDS2 phenotype, and twelve genes from the keratoconus type 7 locus. Because our patient presented with keratoconus, we propose she further refines disease genes at this locus. Among previously suggested candidates, we exclude DOCK9 and STK24, and propose as best candidates IPO5, DNAJC3, MBNL2 and RAP2A. In conclusion, we report a novel phenotypic association of Feingold syndrome type 2 and keratoconus, a likely contiguous gene syndrome due to a large genomic deletion on 13q spanning MIR17HG and a still to be identified gene for keratoconus.
Neurobehavioral Alterations in a Genetic Murine Model of Feingold Syndrome 2.
Feingold syndrome (FS) is an autosomal dominant disorder characterized by microcephaly, short stature, digital anomalies, esophageal/duodenal atresia, facial dysmorphism, and various learning disabilities. Heterozygous deletion of the miR-17-92 cluster is responsible for a subset of FS (Feingold syndrome type 2, FS2), and the developmental abnormalities that characterize this disorder are partially recapitulated in mice that harbor a heterozygous deletion of this cluster (miR-17-92∆/+ mice). Although Feingold patients develop a wide array of learning disabilities, no scientific description of learning/cognitive disabilities, intellectual deficiency, and brain alterations have been described in humans and animal models of FS2. The aim of this study was to draw a behavioral profile, during development and in adulthood, of miR-17-92∆/+ mice, a genetic mouse model of FS2. Moreover, dopamine, norepinephrine and serotonin tissue levels in the medial prefrontal cortex (mpFC), and Hippocampus (Hip) of miR-17-92∆/+ mice were analyzed.Our data showed decreased body growth and reduced vocalization during development. Moreover, selective deficits in spatial ability, social novelty recognition and memory span were evident in adult miR-17-92∆/+ mice compared with healthy controls (WT). Finally, we found altered dopamine as well as serotonin tissue levels, in the mpFC and Hip, respectively, of miR-17-92∆/+ in comparison with WT mice, thus suggesting a possible link between cognitive deficits and altered brain neurotransmission.
Publicações recentes
First Patient Diagnosed as Feingold Syndrome Type 2 with Alport Syndrome and Review of the Current Literature.
Feingold syndrome type 2 in a patient from China.
Distinct molecular pathways mediate Mycn and Myc-regulated miR-17-92 microRNA action in Feingold syndrome mouse models.
A case of Feingold type 2 syndrome associated with keratoconus refines keratoconus type 7 locus on chromosome 13q.
Neurobehavioral Alterations in a Genetic Murine Model of Feingold Syndrome 2.
📚 EuropePMC48 artigos no totalmostrando 5
First Patient Diagnosed as Feingold Syndrome Type 2 with Alport Syndrome and Review of the Current Literature.
Molecular syndromologyFeingold syndrome type 2 in a patient from China.
American journal of medical genetics. Part ADistinct molecular pathways mediate Mycn and Myc-regulated miR-17-92 microRNA action in Feingold syndrome mouse models.
Nature communicationsA case of Feingold type 2 syndrome associated with keratoconus refines keratoconus type 7 locus on chromosome 13q.
European journal of medical geneticsNeurobehavioral Alterations in a Genetic Murine Model of Feingold Syndrome 2.
Behavior geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de Feingold, tipo 2.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de Feingold, tipo 2
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- First Patient Diagnosed as Feingold Syndrome Type 2 with Alport Syndrome and Review of the Current Literature.
- Feingold syndrome type 2 in a patient from China.
- Distinct molecular pathways mediate Mycn and Myc-regulated miR-17-92 microRNA action in Feingold syndrome mouse models.
- A case of Feingold type 2 syndrome associated with keratoconus refines keratoconus type 7 locus on chromosome 13q.
- Neurobehavioral Alterations in a Genetic Murine Model of Feingold Syndrome 2.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:391646(Orphanet)
- OMIM OMIM:614326(OMIM)
- MONDO:0013691(MONDO)
- GARD:17625(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome de Feingold, tipo 2
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)