A Cistinose Nefropática Infantil é a forma mais comum e grave de cistinose. É uma doença que afeta o metabolismo do corpo, causada pelo acúmulo de uma substância chamada cistina dentro das células, em estruturas que funcionam como pequenas "lixeiras" celulares, chamadas lisossomos. Esse acúmulo provoca danos em diversos órgãos e tecidos, especialmente nos rins e nos olhos.
Introdução
O que você precisa saber de cara
A Cistinose Nefropática Infantil é a forma mais comum e grave de cistinose. É uma doença que afeta o metabolismo do corpo, causada pelo acúmulo de uma substância chamada cistina dentro das células, em estruturas que funcionam como pequenas "lixeiras" celulares, chamadas lisossomos. Esse acúmulo provoca danos em diversos órgãos e tecidos, especialmente nos rins e nos olhos.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 12 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 28 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Cystine/H(+) symporter that mediates export of cystine, the oxidized dimer of cysteine, from lysosomes (PubMed:11689434, PubMed:15128704, PubMed:18337546, PubMed:22232659, PubMed:29467429, PubMed:33208952, PubMed:36113465). Plays an important role in melanin synthesis by catalyzing cystine export from melanosomes, possibly by inhibiting pheomelanin synthesis (PubMed:22649030). In addition to cystine export, also acts as a positive regulator of mTORC1 signaling in kidney proximal tubular cells, v
Lysosome membraneMelanosome membraneCell membrane
Cystinosis, nephropathic type
A form of cystinosis, a lysosomal storage disease due to defective transport of cystine across the lysosomal membrane. This results in cystine accumulation and crystallization in the cells causing widespread tissue damage. The classical nephropathic form has onset in the first year of life and is characterized by a polyuro-polydipsic syndrome, marked height-weight growth delay, generalized impaired proximal tubular reabsorptive capacity, with severe fluid-electrolyte balance alterations, renal failure, ocular symptoms and other systemic complications.
Variantes genéticas (ClinVar)
702 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 3 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Cistinose nefropática da infância
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
Ocular manifestations and multimodal imaging in infantile nephropathic cystinosis.
Neuroretinal structure changes in infantile nephropathic cystinosis.
The aim of this study was to investigate the neuroretinal structure of patients with the lysosomal storage disease cystinosis. In this retrospective cross-sectional analysis, optical coherence tomography (OCT) was used to measure the peripapillary retinal nerve fiber layer (pRNFL), the optic disc volumes, the prelaminar depth and the macular ganglion cell layer volumes (mGCL) in patients with genetically confirmed infantile nephropathic cystinosis. The same measurements were repeated in an age -and spherical equivalent (SE) matched, healthy control group. The cystinosis group included 40 patients (40 eyes) with a mean age of 20.6 ± 8.6 years and a SE of 0.47 ± 1.85. The healthy control group consisted of 30 patients (30 eyes) with a mean age of 20.7 ± 12.5 years and a SE of 0.47 ± 1.29. A pronounced deposition of crystals in the optic disc was observed in all cystinosis cases. Cystine crystals follow the nerve fibers in a dense, pearl-string pattern. A significantly thicker pRNFL and a higher rate of positive prelaminar depth was evident in the cystinosis group (839.7 ± 151.0 μm vs. 775.7 ± 79.6 μm, p = 0.004). A significantly smaller mGCL volume was found in the cystinosis group as compared to normal controls (0.25 ± 0.03 mm³ vs. 0.35 ± 0.03 mm³, p = 0.036). Cystinosis leads to pronounced crystal accumulation in the optic disc in early stages of the disease. This accumulation occurs in concomitance with the well-described cystine crystal deposits in the cornea, which have previously been considered the foremost ocular sign of cystinosis. The pearl-string appearance of crystal deposition suggests a primarily glial localization. A significantly thicker pRNFL and a higher rate of positive prelaminar depth was observed in the OCT scans of cystinosis patients, explaining the clinical impression of a crowded optic disc. Additionally, retinal neurodegeneration was significant in patients with cystinosis if compared to healthy controls. The optic disc crowding may result from the dense deposition of cystine crystals in the optic nerve head and the GCL thinning could be due to metabolically induced ganglion cell atrophy. However, the exact reason for these changes remains to be elucidated.
Diagnosis and management of cystinosis: systematic review for a clinical practice guideline.
Cystinosis is a rare genetic disorder, with the majority of patients suffering from infantile nephropathic cystinosis, the most severe form. If left untreated, cystinosis causes serious morbidity, initially through progressive kidney and eye disease, followed by systemic and multiorgan involvement, ultimately leading to premature death. In this systematic review, we summarize the evidence for cystinosis to support the development of an evidence-based clinical practice guideline (SELECT - S3 guideline for cystinosis). We searched MEDLINE, Embase, CENTRAL, CINAHL, and other databases for relevant studies published from 1980 onward. We screened literature dually and independently for eligibility. Primary researchers extracted data, rated the risk of bias of included studies, and rated the certainty of the evidence (CoE). Secondary researchers reviewed for completeness and accuracy. We applied a staggered approach, prioritizing available controlled studies and synthesizing results narratively. We considered 56 studies in our synthesis and assessed findings relevant to 17 of 31 key questions. We identified evidence for 62 of 213 outcomes. Fifty-two outcomes had very low CoE, three low, four moderate, and three high. The moderate and high CoE findings came from indirect comparisons (other chronic multi-organ diseases). There was low evidence that delayed-release cysteamine therapy makes little to no difference on cystine levels compared to immediate-release cysteamine therapy; however, delayed-release cysteamine was associated with a slight increase in adverse events. Starting systemic cysteamine treatment early likely improves renal function compared to a later start. Most included studies lacked a control group, had a high risk of bias, and had a small sample size. Evidence informing the diagnosis and management of cystinosis is limited, with most findings rated as very low certainty. Few direct comparisons involving only individuals with cystinosis yielded low certainty findings, while moderate to high certainty evidence was found only in indirect comparisons. These findings underscore a critical challenge in managing cystinosis: the balancing act of clinical decision-making in the context of lacking evidence. Nonetheless, this systematic review synthesized the best available data for a clinical practice guideline and highlighted specific areas where future research could strengthen the evidence base. The online version contains supplementary material available at 10.1186/s13023-025-03974-z.
Local Guidance on the Management of Nephropathic Cystinosis in the Gulf Cooperation Council (GCC) Region.
Cystinosis is a rare systemic disease characterized by the accumulation of cystine in tissues, leading to multi-organ damage. Infantile nephropathic cystinosis is the dominant and severe form of cystinosis with critical renal manifestations that require kidney transplantation at an early age if left untreated. Cysteamine, the lifelong cystine-depleting therapy, is the mainstay treatment of nephropathic cystinosis. Cysteamine prevents cystine crystal formation and delays disease progression. While the initially introduced cysteamine consists of an immediate-release (IR) formulation, a delayed-release (DR) formulation has been developed with a simplified dosing regimen (Q12H instead of Q6H) and an improved quality of life while maintaining comparable efficacy. Due to the rare incidence of the disease and lack of international guidelines, diagnosis and treatment initiation are oftentimes delayed, leading to a poor prognosis. Pediatric and adult nephrologists from Kuwait, Saudi Arabia, the United Arab Emirates (UAE), and Qatar, in addition to one international expert from Amsterdam, convened to share their clinical experience, reflecting on the challenges encountered and therapeutic approaches followed in the management of nephropathic cystinosis in the Gulf Cooperation Council (GCC) region. Experts completed a multiple-choice questionnaire and engaged in structured discussions, where they shed light on gaps and limitations with regard to diagnostic tests and criteria to ensure early diagnosis and timely treatment initiation. Based on available literature, experts suggested an algorithm to help guide nephropathic cystinosis management in the GCC. It is highly recommended for patients who do not tolerate IR-cysteamine and do not adhere to IR-cysteamine treatment to switch to DR-cysteamine. Given the systemic nature of the disease, a multi-disciplinary approach is required for optimal disease management.
Cystinosis metabolic bone disease: inflammatory profile in human peripheral blood mononuclear cells and derived osteoclasts.
Cystinosis metabolic bone disease (CMBD) is an emerging concept in infantile nephropathic cystinosis, patients presenting with bone pains, fractures, and deformations during teenage or early adulthood. The underlying mechanisms remain unclear. Our aim was to explore the pro-inflammatory profile of osteoclastic lineage in cystinotic patients. We obtained blood samples from 14 cystinotic patients and 10 pediatric healthy controls. Peripheral blood mononuclear cells (PBMCs) were isolated and used to explore by RT-qPCR the transcript expression of 8 inflammatory markers (Il-6, Il-8, Il-1β, CXCL1, CCL2/MCP-1, CXCR3, Il-1 Receptor, Il-6 Receptor). In addition, when possible, PBMCs were differentiated into osteoclasts for further experiments. The expression of Il-6, IL-8, CXCR3, and CCL2/MCP-1 was significantly increased in PBMCs from cystinotic patients. We also explored the expression of Il-1 Receptor and Il-6 Receptor, two major pro-osteoclastic signal inducers, in osteoclasts differentiated from PBMCs from controls (N = 3) and patients (N = 4). The expression of IL-1 Receptor (but not IL-6 receptor) was increased in osteoclasts obtained from cystinotic patients. There is an inflammatory profile in PBMCs and osteoclastic lineage in cells obtained from cystinotic patients. CXCR3 and MCP-1 stimulate migration and activation of macrophages, that may explain the previously reported local increased osteoclastogenesis. The osteoclastic overexpression of IL-1 Receptor is a relevant observation in the field since blocking Il-1β signaling has recently been proposed as a novel therapeutic approach to improve muscular wasting in this orphan disease. • Cystinosis metabolic bone disease (CMBD), an emerging concept with unclear underlying mechanisms, induces bone pains, fractures and deformations in patients with cystinosis. • Blocking Il-1β signaling may be a novel therapeutic approach to improve muscular wasting in cystinosis. • There is an inflammatory profile in PBMCs and osteoclastic lineage in cells obtained from cystinotic patients, with an over-expression of IL-1 Receptor in osteoclasts. • We provide another experimental rationale to propose targeted anti-inflammatory therapies in cystinotic patients with severe bone disease.
Publicações recentes
Neuroretinal structure changes in infantile nephropathic cystinosis.
Diagnosis and management of cystinosis: systematic review for a clinical practice guideline.
Local Guidance on the Management of Nephropathic Cystinosis in the Gulf Cooperation Council (GCC) Region.
Ocular manifestations and multimodal imaging in infantile nephropathic cystinosis.
Cystinosis metabolic bone disease: inflammatory profile in human peripheral blood mononuclear cells and derived osteoclasts.
📚 EuropePMC55 artigos no totalmostrando 56
Neuroretinal structure changes in infantile nephropathic cystinosis.
Orphanet journal of rare diseasesDiagnosis and management of cystinosis: systematic review for a clinical practice guideline.
Orphanet journal of rare diseasesLocal Guidance on the Management of Nephropathic Cystinosis in the Gulf Cooperation Council (GCC) Region.
Children (Basel, Switzerland)Ocular manifestations and multimodal imaging in infantile nephropathic cystinosis.
QJM : monthly journal of the Association of PhysiciansCystinosis metabolic bone disease: inflammatory profile in human peripheral blood mononuclear cells and derived osteoclasts.
European journal of pediatricsLeptin signalling altered in infantile nephropathic cystinosis-related bone disorder.
Journal of cachexia, sarcopenia and muscleOcular Involvement in Patients with Infantile Nephropathic Cystinosis.
Turkish journal of ophthalmologyLong-term effects of luteolin in a mouse model of nephropathic cystinosis.
Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapieMorphological changes and their associations with clinical parameters in children with nephropathic cystinosis and chronic kidney disease prior to kidney replacement therapy over 25 years.
Pediatric nephrology (Berlin, Germany)Renal transplantation for infantile and juvenile cystinosis: Two case report and review of the literature.
Transplant immunologyChest configuration in children and adolescents with infantile nephropathic cystinosis compared with other chronic kidney disease entities and its clinical determinants.
Pediatric nephrology (Berlin, Germany)Posterior Segment Involvement in Infantile Nephropathic Cystinosis - A Review.
Klinische Monatsblatter fur AugenheilkundeCorneal Manifestation in Patients with Infantile Nephropathic Cystinosis.
Klinische Monatsblatter fur AugenheilkundeMetabolic Advantage of 25(OH)D3 versus 1,25(OH)2D3 Supplementation in Infantile Nephropathic Cystinosis-Associated Adipose Tissue Browning and Muscle Wasting.
CellsOutcome of infantile nephropathic cystinosis depends on early intervention, not genotype: A multicenter sibling cohort study.
Journal of inherited metabolic diseaseBeneficial effects of starting oral cysteamine treatment in the first 2 months of life on glomerular and tubular kidney function in infantile nephropathic cystinosis.
Molecular genetics and metabolismRelationship between age at initiation of cysteamine treatment, adherence with therapy, and glomerular kidney function in infantile nephropathic cystinosis.
Molecular genetics and metabolismPatients With Infantile Nephropathic Cystinosis in Germany and Austria: A Retrospective Cohort Study.
Frontiers in medicineClinical and genetic characteristics of Tunisian children with infantile nephropathic cystinosis.
Pediatric nephrology (Berlin, Germany)Newborn Screening: Review of its Impact for Cystinosis.
CellsAnalysis of tear film in cystinosis patients treated with topical viscous cysteamine hydrochloride (Cystadrops®).
European journal of ophthalmologyMuscle and Bone Impairment in Infantile Nephropathic Cystinosis: New Concepts.
CellsBody growth, upper arm fat area, and clinical parameters in children with nephropathic cystinosis compared with other pediatric chronic kidney disease entities.
Journal of inherited metabolic diseaseNeuromuscular conditions and the impact of cystine-depleting therapy in infantile nephropathic cystinosis: A cross-sectional analysis of 55 patients.
Journal of inherited metabolic diseaseSpectral domain optical coherence tomography-based retinochoroidal cystine crystal score: a window into infantile nephropathic cystinosis.
The British journal of ophthalmologyA Leptin Receptor Antagonist Attenuates Adipose Tissue Browning and Muscle Wasting in Infantile Nephropathic Cystinosis-Associated Cachexia.
CellsTargeting interleukin-1 for reversing fat browning and muscle wasting in infantile nephropathic cystinosis.
Journal of cachexia, sarcopenia and muscleMore than tubular dysfunction: cystinosis and kidney outcomes.
Journal of nephrologyTesticular function in males with infantile nephropathic cystinosis.
Human reproduction (Oxford, England)Posterior segment optical coherence tomography findings in a case of nephropathic cystinosis.
Saudi journal of ophthalmology : official journal of the Saudi Ophthalmological SocietyThe Importance of the Early Diagnosis of Infantile Nephropathic Cystinosis: A Case Report.
Klinische PadiatrieA severe course of serogroup W meningococcemia in a patient with infantile nephropathic cystinosis.
Human vaccines & immunotherapeuticsVitamin D repletion ameliorates adipose tissue browning and muscle wasting in infantile nephropathic cystinosis-associated cachexia.
Journal of cachexia, sarcopenia and muscleCTNS mRNA molecular analysis revealed a novel mutation in a child with infantile nephropathic cystinosis: a case report.
BMC nephrologyOral alterations in patients with cystinosis.
Special care in dentistry : official publication of the American Association of Hospital Dentists, the Academy of Dentistry for the Handicapped, and the American Society for Geriatric DentistryAsymmetrical Ocular Manifestations of Nephropathic Cystinosis; A Case Report.
The American journal of case reportsTherapeutic Problems and Pregnancy in a Patient With Infantile Nephropathic Cystinosis: A Case Report.
Transplantation proceedingsThe Ocular Status of Cystinosis Patients Receiving a Hospital Pharmacy-Made Preparation of Cysteamine Eye Drops: A Case Series.
Ophthalmology and therapyLatest Clinical Approaches in the Ocular Management of Cystinosis: A Review of Current Practice and Opinion from the Ophthalmology Cystinosis Forum.
Ophthalmology and therapyAtypical onset of nephropathic infantile cystinosis in a Russian patient with rare CTNS mutation.
Clinical case reportsNephropathic Cystinosis: Symptoms, Treatment, and Perspectives of a Systemic Disease.
Frontiers in pediatricsSlow progression of renal failure in a child with infantile cystinosis.
CEN case reportsEffects of long-term cysteamine treatment in patients with cystinosis.
Pediatric nephrology (Berlin, Germany)Mutation analysis of the CTNS gene in Iranian patients with infantile nephropathic cystinosis: identification of two novel mutations.
Human genome variationThe Clinical and Mutational Spectrum of Turkish Patients with Cystinosis.
Clinical journal of the American Society of Nephrology : CJASNInfantile Nephropathic Cystinosis: A Novel CTNS Mutation.
The Eurasian journal of medicineFirst Successful Conception Induced by a Male Cystinosis Patient.
JIMD reportsOcular Complications of Infantile Nephropathic Cystinosis.
The Journal of pediatricsValue of Renal Biopsy in Diagnosing Infantile Nephropathic Cystinosis Associated With Secondary Nephrogenic Diabetes Insipidus.
Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology SocietyMuscle wasting and adipose tissue browning in infantile nephropathic cystinosis.
Journal of cachexia, sarcopenia and muscleInfantile nephropathic cystinosis with incomplete fanconi syndrome, hypothyroidism, hydro-uretero-nephrosis, and megacystis.
Saudi journal of kidney diseases and transplantation : an official publication of the Saudi Center for Organ Transplantation, Saudi Arabia[COMPARISON OF FREE CARNITINE LEVELS WITH NUTRITIONAL STATUS IN INFANTILE NEPHROPATHYC CISTINOSIS PATIENTS].
Nutricion hospitalariaMolecular analysis of the CTNS gene in Jordanian families with nephropathic cystinosis.
Nefrologia : publicacion oficial de la Sociedad Espanola NefrologiaCTNS mutations in publicly-available human cystinosis cell lines.
Molecular genetics and metabolism reportsHirschsprung's disease with infantile nephropathic cystinosis.
Journal of Indian Association of Pediatric SurgeonsA mouse model suggests two mechanisms for thyroid alterations in infantile cystinosis: decreased thyroglobulin synthesis due to endoplasmic reticulum stress/unfolded protein response and impaired lysosomal processing.
EndocrinologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Cistinose nefropática da infância.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Cistinose nefropática da infância
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Ocular manifestations and multimodal imaging in infantile nephropathic cystinosis.
- Neuroretinal structure changes in infantile nephropathic cystinosis.
- Diagnosis and management of cystinosis: systematic review for a clinical practice guideline.
- Local Guidance on the Management of Nephropathic Cystinosis in the Gulf Cooperation Council (GCC) Region.
- Cystinosis metabolic bone disease: inflammatory profile in human peripheral blood mononuclear cells and derived osteoclasts.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:411629(Orphanet)
- MONDO:0018467(MONDO)
- GARD:9755(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q56014157(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
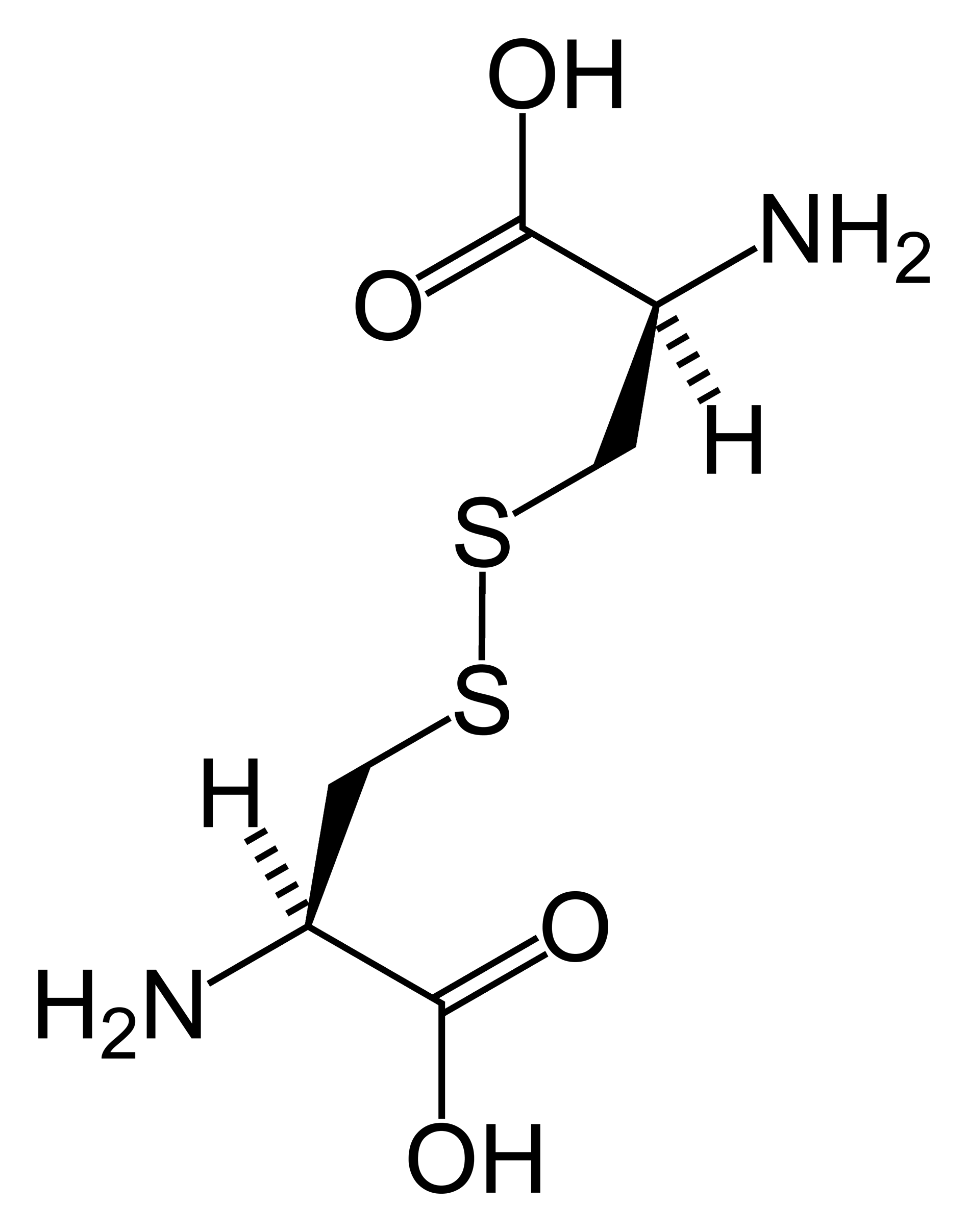
Cistinose nefropática da infância
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Reposicionamento
- fonte: Drug Repurposing Hub
- Ensaios clínicos
- fonte: ClinicalTrials.gov