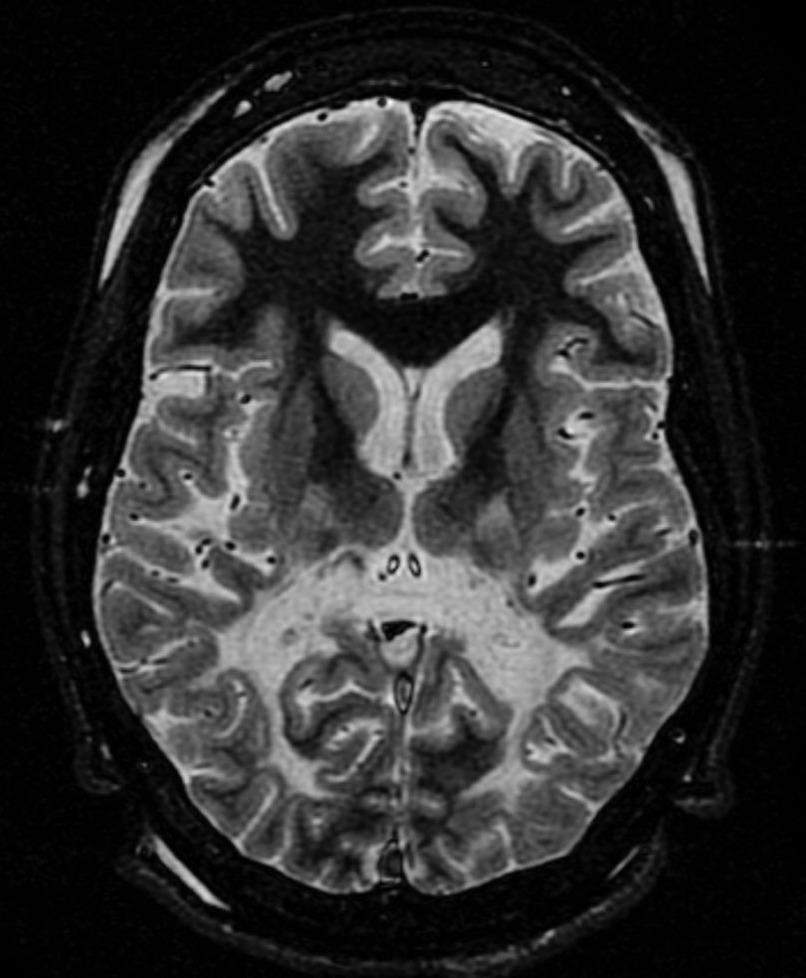
Leucodistrofias são um grupo de doenças genéticas raras, progressivas e metabólicas que afetam o cérebro, a medula espinhal e, muitas vezes, os nervos periféricos. Cada tipo de leucodistrofia é causado por uma alteração genética específica que leva ao desenvolvimento anormal ou à destruição da substância branca (a bainha de mielina) do cérebro. A bainha de mielina é o revestimento protetor dos nervos e, sem ela, os nervos não conseguem funcionar normalmente. Cada tipo de leucodistrofia afeta uma parte diferente da bainha de mielina, o que leva a uma variedade de problemas neurológicos.
Introdução
O que você precisa saber de cara
Leucodistrofias são um grupo de doenças genéticas raras, progressivas e metabólicas que afetam o cérebro, a medula espinhal e, muitas vezes, os nervos periféricos. Cada tipo de leucodistrofia é causado por uma alteração genética específica que leva ao desenvolvimento anormal ou à destruição da substância branca (a bainha de mielina) do cérebro. A bainha de mielina é o revestimento protetor dos nervos e, sem ela, os nervos não conseguem funcionar normalmente. Cada tipo de leucodistrofia afeta uma parte diferente da bainha de mielina, o que leva a uma variedade de problemas neurológicos.
Tem tratamento?
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 510 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 1152 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
62 genes identificados com associação a esta condição.
Catalyzes the specific attachment of an amino acid to its cognate tRNA in a 2 step reaction: the amino acid (AA) is first activated by ATP to form AA-AMP and then transferred to the acceptor end of the tRNA
Cytoplasm, cytosol
Hypomyelination with brainstem and spinal cord involvement and leg spasticity
An autosomal recessive leukoencephalopathy characterized by onset in the first year of life of severe spasticity, mainly affecting the lower limbs and resulting in an inability to achieve independent ambulation. Affected individuals show delayed motor development and nystagmus; some may have mild intellectual disability. Brain MRI shows hypomyelination and white matter lesions in the cerebrum, brainstem, cerebellum, and spinal cord.
Accessory subunit of the mitochondrial membrane respiratory chain NADH dehydrogenase (Complex I), that is believed not to be involved in catalysis. Complex I functions in the transfer of electrons from NADH to the respiratory chain. The immediate electron acceptor for the enzyme is believed to be ubiquinone
Mitochondrion inner membrane
Mitochondrial complex I deficiency, nuclear type 13
A form of mitochondrial complex I deficiency, the most common biochemical signature of mitochondrial disorders, a group of highly heterogeneous conditions characterized by defective oxidative phosphorylation, which collectively affects 1 in 5-10000 live births. Clinical disorders have variable severity, ranging from lethal neonatal disease to adult-onset neurodegenerative disorders. Phenotypes include macrocephaly with progressive leukodystrophy, non-specific encephalopathy, cardiomyopathy, myopathy, liver disease, Leigh syndrome, Leber hereditary optic neuropathy, and some forms of Parkinson disease. MC1DN13 transmission pattern is consistent with autosomal recessive inheritance.
Transcription factor with repressor activity involved in the regulation of axon-glial interactions at myelin paranodes in oligodendrocytes. Binds to the consensus DNA sequence 5'-(A/T)TTAATGA-3'. In oligodendrocytes, binds to MBP and PLP1 promoter regions
Nucleus
Spastic ataxia 8, autosomal recessive, with hypomyelinating leukodystrophy
An autosomal recessive neurodegenerative disorder characterized by early-onset hypotonia which progresses to a pyramidal syndrome with ataxia, spasticity, hyperreflexia, weakness and loss of ambulation. Brain imaging shows cerebellar atrophy and hypomyelinating leukodystrophy.
Major component of the axial/lateral elements of synaptonemal complexes (SCS) during meiotic prophase. Plays a role in the assembly of synaptonemal complexes. Required for normal meiotic chromosome synapsis during oocyte and spermatocyte development and for normal male and female fertility. Required for insertion of SYCP3 into synaptonemal complexes. May be involved in the organization of chromatin by temporarily binding to DNA scaffold attachment regions. Requires SYCP3, but not SYCP1, in order
NucleusChromosome
Spermatogenic failure 1
An infertility disorder characterized by azoospermia due to spermatogenic arrest during meiosis. Meiotic arrest is characterized by germ cells that enter meiosis and undergo the first chromosomal reduction from 4n to 2n, but that are then unable to proceed further. This results in tubules containing spermatocytes as the latest developmental stage of germ cells. Meiotically arrested spermatocytes accumulate in the tubules and degenerate. Both autosomal recessive and autosomal dominant inheritance have been reported.
Catalyzes the oxidation of medium and long chain aliphatic aldehydes to fatty acids. Active on a variety of saturated and unsaturated aliphatic aldehydes between 6 and 24 carbons in length (PubMed:18035827, PubMed:18182499, PubMed:22633490, PubMed:25047030, PubMed:9133646, PubMed:9662422). Responsible for conversion of the sphingosine 1-phosphate (S1P) degradation product hexadecenal to hexadecenoic acid (PubMed:22633490)
Microsome membraneEndoplasmic reticulum membrane
Sjoegren-Larsson syndrome
An autosomal recessive neurocutaneous disorder characterized by a combination of severe intellectual disability, spastic di- or tetraplegia and congenital ichthyosis. Ichthyosis is usually evident at birth with varying degrees of erythema and scaling, neurologic symptoms appear in the first or second year of life. Most patients have an IQ of less than 60. Additional clinical features include glistening white spots on the retina, seizures, short stature and speech defects.
Mitochondrial NAD(+) kinase that phosphorylates NAD(+) to yield NADP(+). Can use both ATP or inorganic polyphosphate as the phosphoryl donor. Also has weak NADH kinase activity in vitro; however NADH kinase activity is much weaker than the NAD(+) kinase activity and may not be relevant in vivo
Mitochondrion
2,4-dienoyl-CoA reductase deficiency
A rare, autosomal recessive, inborn error of polyunsaturated fatty acids and lysine metabolism, resulting in mitochondrial dysfunction. Affected individuals have a severe encephalopathy with neurologic and metabolic abnormalities beginning in early infancy. Laboratory studies show increased C10:2 carnitine levels and hyperlysinemia.
Acts as a component of the translation initiation factor 2B (eIF2B) complex, which catalyzes the exchange of GDP for GTP on eukaryotic initiation factor 2 (eIF2) gamma subunit (PubMed:25858979, PubMed:27023709, PubMed:31048492). Its guanine nucleotide exchange factor activity is repressed when bound to eIF2 complex phosphorylated on the alpha subunit, thereby limiting the amount of methionyl-initiator methionine tRNA available to the ribosome and consequently global translation is repressed (Pub
Cytoplasm, cytosol
Leukoencephalopathy with vanishing white matter 4
An autosomal recessive brain disease characterized by neurological features including progressive cerebellar ataxia, spasticity, and cognitive deficits. Brain imaging shows abnormal white matter that vanishes over time and is replaced by cerebrospinal fluid. Disease severity ranges from fatal infantile forms to adult forms without neurological deterioration. The disease is progressive with, in most individuals, additional episodes of rapid deterioration following febrile infections or minor head trauma. Death may occurs after a variable period after disease onset, usually following an episode of fever and coma. A subset of affected females with milder forms of the disease who survive to adolescence exhibit ovarian dysfunction. This variant of the disorder is called ovarioleukodystrophy.
Transmembrane protein mainly expressed in brain astrocytes that may play a role in transport across the blood-brain and brain-cerebrospinal fluid barriers (PubMed:22328087). Regulates the response of astrocytes to hypo-osmosis by promoting calcium influx (PubMed:22328087). May function as regulatory protein of membrane protein complexes such as ion channels (Probable)
MembraneCell membraneCytoplasm, perinuclear regionEndoplasmic reticulum
Megalencephalic leukoencephalopathy with subcortical cysts 1
A syndrome of cerebral leukoencephalopathy and megalencephaly characterized by ataxia, spasticity, seizures, delay in motor development and mild intellectual disability. The brain appears swollen on magnetic resonance imaging, with diffuse white-matter abnormalities and the invariable presence of subcortical cysts in frontal and temporal lobes.
Involved in peroxisomal proliferation (PubMed:9792670). May regulate peroxisome division by recruiting the dynamin-related GTPase DNM1L to the peroxisomal membrane (PubMed:12618434). Promotes membrane protrusion and elongation on the peroxisomal surface (PubMed:20826455)
Peroxisome membrane
Peroxisome biogenesis disorder 14B
An autosomal recessive peroxisome biogenesis disorder characterized clinically by mild intellectual disability, congenital cataracts, progressive hearing loss, and polyneuropathy. Additionally, recurrent migraine-like episodes following mental stress or physical exertion, not a common feature in peroxisome disorders, are observed.
Necessary for early peroxisomal biogenesis. Acts both as a cytosolic chaperone and as an import receptor for peroxisomal membrane proteins (PMPs). Binds and stabilizes newly synthesized PMPs in the cytoplasm by interacting with their hydrophobic membrane-spanning domains, and targets them to the peroxisome membrane by binding to the integral membrane protein PEX3. Excludes CDKN2A from the nucleus and prevents its interaction with MDM2, which results in active degradation of TP53
CytoplasmPeroxisome membrane
Peroxisome biogenesis disorder complementation group 14
A peroxisomal disorder arising from a failure of protein import into the peroxisomal membrane or matrix. The peroxisome biogenesis disorders (PBD group) are genetically heterogeneous with at least 14 distinct genetic groups as concluded from complementation studies. Include disorders are: Zellweger syndrome (ZWS), neonatal adrenoleukodystrophy (NALD), infantile Refsum disease (IRD), and classical rhizomelic chondrodysplasia punctata (RCDP). ZWS, NALD and IRD are distinct from RCDP and constitute a clinical continuum of overlapping phenotypes known as the Zellweger spectrum (PBD-ZSS).
Catalyzes the deacetylation of N-acetylaspartic acid (NAA) to produce acetate and L-aspartate. NAA occurs in high concentration in brain and its hydrolysis NAA plays a significant part in the maintenance of intact white matter. In other tissues it acts as a scavenger of NAA from body fluids
CytoplasmNucleus
Canavan disease
A rare neurodegenerative condition of infancy or childhood characterized by white matter vacuolization and demyelination that gives rise to a spongy appearance. The clinical features are onset in early infancy, atonia of neck muscles, hypotonia, hyperextension of legs and flexion of arms, blindness, severe mental defect, megalocephaly, and death by 18 months on the average.
May be involved in vesicular trafficking from the Golgi apparatus to the cell membrane. Plays a role in the maintenance of the myelin sheath, and in axon-glia and glia-glia interactions
MembraneCell membrane
Leukodystrophy, hypomyelinating, 28
A form of hypomyelinating leukodystrophy, a group of heterogeneous disorders characterized by persistent deficit of myelin observed on brain imaging. HLD28 is an autosomal recessive form characterized by developmental delay and nystagmus in infancy, followed by significant learning disabilities and progressive motor deterioration within the first decade.
Component of the U7 snRNP complex that is involved in the histone 3'-end pre-mRNA processing (PubMed:11574479, PubMed:16914750, PubMed:33230297). Increases U7 snRNA levels but not histone 3'-end pre-mRNA processing activity, when overexpressed (PubMed:11574479, PubMed:16914750). Required for cell cycle progression from G1 to S phases (By similarity). Binds specifically to the Sm-binding site of U7 snRNA (PubMed:11574479, PubMed:16914750)
Nucleus
Aicardi-Goutieres syndrome 8
A form of Aicardi-Goutieres syndrome, a genetically heterogeneous disease characterized by cerebral atrophy, leukoencephalopathy, intracranial calcifications, chronic cerebrospinal fluid (CSF) lymphocytosis, increased CSF alpha-interferon, and negative serologic investigations for common prenatal infection. Clinical features as thrombocytopenia, hepatosplenomegaly and elevated hepatic transaminases along with intermittent fever may erroneously suggest an infective process. Severe neurological dysfunctions manifest in infancy as progressive microcephaly, spasticity, dystonic posturing and profound psychomotor retardation. Death often occurs in early childhood. AGS8 inheritance is autosomal recessive.
Major cellular 3'-to-5' DNA exonuclease which digests single-stranded DNA (ssDNA) and double-stranded DNA (dsDNA) with mismatched 3' termini (PubMed:10391904, PubMed:10393201, PubMed:17293595). Prevents cell-intrinsic initiation of autoimmunity (PubMed:10391904, PubMed:10393201, PubMed:17293595). Acts by metabolizing DNA fragments from endogenous retroelements, including L1, LTR and SINE elements (PubMed:10391904, PubMed:10393201, PubMed:17293595). Plays a key role in degradation of DNA fragment
NucleusCytoplasm, cytosolEndoplasmic reticulum membrane
Aicardi-Goutieres syndrome 1
A form of Aicardi-Goutieres syndrome, a genetically heterogeneous disease characterized by cerebral atrophy, leukoencephalopathy, intracranial calcifications, chronic cerebrospinal fluid (CSF) lymphocytosis, increased CSF alpha-interferon, and negative serologic investigations for common prenatal infection. Clinical features as thrombocytopenia, hepatosplenomegaly and elevated hepatic transaminases along with intermittent fever may erroneously suggest an infective process. Severe neurological dysfunctions manifest in infancy as progressive microcephaly, spasticity, dystonic posturing and profound psychomotor retardation. Death often occurs in early childhood.
Non catalytic subunit of RNase H2, an endonuclease that specifically degrades the RNA of RNA:DNA hybrids. Participates in DNA replication, possibly by mediating the removal of lagging-strand Okazaki fragment RNA primers during DNA replication. Mediates the excision of single ribonucleotides from DNA:RNA duplexes
Nucleus
Aicardi-Goutieres syndrome 3
A form of Aicardi-Goutieres syndrome, a genetically heterogeneous disease characterized by cerebral atrophy, leukoencephalopathy, intracranial calcifications, chronic cerebrospinal fluid (CSF) lymphocytosis, increased CSF alpha-interferon, and negative serologic investigations for common prenatal infection. Clinical features as thrombocytopenia, hepatosplenomegaly and elevated hepatic transaminases along with intermittent fever may erroneously suggest an infective process. Severe neurological dysfunctions manifest in infancy as progressive microcephaly, spasticity, dystonic posturing and profound psychomotor retardation. Death often occurs in early childhood.
GFAP, a class-III intermediate filament, is a cell-specific marker that, during the development of the central nervous system, distinguishes astrocytes from other glial cells
Cytoplasm
Alexander disease
A rare disorder of the central nervous system. The most common form affects infants and young children, and is characterized by progressive failure of central myelination, usually leading to death within the first decade. Infants with Alexander disease develop a leukodystrophy with macrocephaly, seizures, and psychomotor retardation. Patients with juvenile or adult forms typically experience ataxia, bulbar signs and spasticity, and a more slowly progressive course. Histologically, Alexander disease is characterized by Rosenthal fibers, homogeneous eosinophilic inclusions in astrocytes.
Acts as a component of the translation initiation factor 2B (eIF2B) complex, which catalyzes the exchange of GDP for GTP on eukaryotic initiation factor 2 (eIF2) gamma subunit (PubMed:25858979, PubMed:27023709, PubMed:31048492). Its guanine nucleotide exchange factor activity is repressed when bound to eIF2 complex phosphorylated on the alpha subunit, thereby limiting the amount of methionyl-initiator methionine tRNA available to the ribosome and consequently global translation is repressed (Pub
Cytoplasm, cytosol
Leukoencephalopathy with vanishing white matter 5
An autosomal recessive brain disease characterized by neurological features including progressive cerebellar ataxia, spasticity, and cognitive deficits. Brain imaging shows abnormal white matter that vanishes over time and is replaced by cerebrospinal fluid. Disease severity ranges from fatal infantile forms to adult forms without neurological deterioration. The disease is progressive with, in most individuals, additional episodes of rapid deterioration following febrile infections or minor head trauma. Death may occurs after a variable period after disease onset, usually following an episode of fever and coma. A subset of affected females with milder forms of the disease who survive to adolescence exhibit ovarian dysfunction. This variant of the disorder is called ovarioleukodystrophy.
Non-catalytic component of the multisynthase complex. Stimulates the catalytic activity of cytoplasmic arginyl-tRNA synthase (PubMed:10358004). Binds tRNA. Possesses inflammatory cytokine activity (PubMed:11306575). Negatively regulates TGF-beta signaling through stabilization of SMURF2 by binding to SMURF2 and inhibiting its SMAD7-mediated degradation (By similarity). Involved in glucose homeostasis through induction of glucagon secretion at low glucose levels (By similarity). Promotes dermal f
NucleusCytoplasm, cytosolSecretedEndoplasmic reticulumGolgi apparatus
Leukodystrophy, hypomyelinating, 3
A severe autosomal recessive hypomyelinating leukodystrophy characterized by early infantile onset of global developmental delay, lack of development, lack of speech acquisition, and peripheral spasticity associated with decreased myelination in the central nervous system.
ATPase required for the post-translational delivery of tail-anchored (TA) proteins to the endoplasmic reticulum (PubMed:17382883). Recognizes and selectively binds the transmembrane domain of TA proteins in the cytosol. This complex then targets to the endoplasmic reticulum by membrane-bound receptors GET1/WRB and CAMLG/GET2, where the tail-anchored protein is released for insertion. This process is regulated by ATP binding and hydrolysis. ATP binding drives the homodimer towards the closed dime
CytoplasmEndoplasmic reticulumNucleus, nucleolus
Cardiomyopathy, dilated, 2H
A form of dilated cardiomyopathy, a disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. CMD2H is an autosomal recessive form characterized by rapid progression and death in early infancy.
Component of a complex required to localize phosphatidylinositol 4-kinase (PI4K) to the plasma membrane (PubMed:26571211). The complex acts as a regulator of phosphatidylinositol 4-phosphate (PtdIns(4)P) synthesis (PubMed:26571211). HYCC1 plays a key role in oligodendrocytes formation, a cell type with expanded plasma membrane that requires generation of PtdIns(4)P (PubMed:26571211). Its role in oligodendrocytes formation probably explains its importance in myelination of the central and periphe
Cytoplasm, cytosolCell membrane
Leukodystrophy, hypomyelinating, 5
A hypomyelinating leukodystrophy associated with congenital cataract. It is clinically characterized by congenital cataract, progressive neurologic impairment, and diffuse myelin deficiency. Affected individuals experience progressive pyramidal and cerebellar dysfunction, muscle weakness and wasting prevailingly in the lower limbs. Mental deficiency ranges from mild to moderate. HLD5 shows clinical variability, but features of hypomyelination combined with increased periventricular white matter water content are consistently observed.
Catalytic core component of RNA polymerase I (Pol I), a DNA-dependent RNA polymerase which synthesizes ribosomal RNA precursors using the four ribonucleoside triphosphates as substrates. Transcribes 47S pre-rRNAs from multicopy rRNA gene clusters, giving rise to 5.8S, 18S and 28S ribosomal RNAs (PubMed:11250903, PubMed:11283244, PubMed:16858408, PubMed:34671025, PubMed:34887565, PubMed:36271492). Pol I-mediated transcription cycle proceeds through transcription initiation, transcription elongati
Nucleus, nucleolusChromosome
Acrofacial dysostosis, Cincinnati type
A form of acrofacial dysostosis, a group of disorders characterized by malformations of the craniofacial skeleton and, in some patients, the limbs. AFDCIN patients may also have structural cardiac defects and neurologic abnormalities including developmental delay, hypotonia, motor delay and seizures. AFDCIN inheritance is autosomal dominant.
Mechanosensitive cation channel with low conductance and high activation threshold (PubMed:30382938, PubMed:31587869, PubMed:37543036). In contrast to TMEM63B, does not show phospholipid scramblase activity (PubMed:39716028). Acts as a regulator of lysosomal morphology by mediating lysosomal mechanosensitivity (By similarity). Important for the baby's first breath and respiration throughout life (PubMed:38127458). Upon lung inflation conducts cation currents in alveolar type 1 and 2 cells trigge
Lysosome membraneEarly endosome membraneCell membrane
Leukodystrophy, hypomyelinating, 19, transient infantile
An autosomal dominant disorder characterized by marked hypomyelination on brain imaging, congenital nystagmus, and motor delay manifesting in early infancy. Both neurologic impairment and abnormal brain imaging spontaneously resolve during childhood.
Has sphingolipid-delta-4-desaturase activity. Converts D-erythro-sphinganine to D-erythro-sphingosine (E-sphing-4-enine) (PubMed:11937514, PubMed:30620337, PubMed:30620338). Catalyzes the equilibrium isomerization of retinols (By similarity)
Mitochondrion membraneEndoplasmic reticulum membrane
Leukodystrophy, hypomyelinating, 18
An autosomal recessive disorder characterized by hypomyelinating leukodystrophy with progressive atrophy of the corpus callosum, thalami and cerebellum, and peripheral neuropathy. Clinical features include very poor psychomotor development, dystonia, severe spasticity, seizures, and failure to thrive.
Required for assembly and stability of the aminoacyl-tRNA synthase complex (PubMed:19131329). Mediates ubiquitination and degradation of FUBP1, a transcriptional activator of MYC, leading to MYC down-regulation which is required for aveolar type II cell differentiation. Blocks MDM2-mediated ubiquitination and degradation of p53/TP53. Functions as a proapoptotic factor
Cytoplasm, cytosolNucleus
Leukodystrophy, hypomyelinating, 17
An autosomal recessive neurodevelopmental disorder characterized by atrophy of cerebral cortex, spinal cord and cerebellum, thin corpus callosum, abnormal signals in the basal ganglia, and features suggesting hypo- or demyelination observed on brain imaging. Clinical manifestations include lack of development, absent speech, microcephaly, spasticity, seizures, and contractures.
Multifunctional protein which primarily functions within the aminoacyl-tRNA synthetase multienzyme complex, also known as multisynthetase complex. Within the complex it catalyzes the attachment of both L-glutamate and L-proline to their cognate tRNAs in a two-step reaction where the amino acid is first activated by ATP to form a covalent intermediate with AMP. Subsequently, the activated amino acid is transferred to the acceptor end of the cognate tRNA to form L-glutamyl-tRNA(Glu) and L-prolyl-t
Cytoplasm, cytosolMembrane
Leukodystrophy, hypomyelinating, 15
An autosomal recessive disorder characterized by hypomyelinating leukodystrophy with thinning of the corpus callosum. Clinical features include motor and cognitive impairment appearing in the first or second decade of life, dystonia, ataxia, spasticity, and dysphagia. Most patients develop severe optic atrophy, and some have hearing loss.
Adapter protein which non-covalently associates with activating receptors found on the surface of a variety of immune cells to mediate signaling and cell activation following ligand binding by the receptors (PubMed:10604985, PubMed:9490415, PubMed:9655483). TYROBP is tyrosine-phosphorylated in the ITAM domain following ligand binding by the associated receptors which leads to activation of additional tyrosine kinases and subsequent cell activation (PubMed:9490415). Also has an inhibitory role in
Cell membrane
Polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy 1
A recessively inherited disease characterized by presenile dementia along with large-scale destruction of cancellous bones. Initial symptoms, starting in the twenties, are pain and swelling resulting from cysts in the wrists and ankles. Extremity bone fractures could occur with minor trauma. At around 30 years of age, patients gradually develop neuropsychiatric symptoms, including epileptic seizures, agnosia, apraxia, speech disorder, memory disturbance, euphoria, and loss of social inhibitions. The disorder usually leads to death in the fifth decade of life.
Peroxisomal docking factor that anchors PEX1 and PEX6 to peroxisome membranes (PubMed:12717447, PubMed:12851857, PubMed:16257970, PubMed:16763195, PubMed:16854980, PubMed:21362118). PEX26 is therefore required for the formation of the PEX1-PEX6 AAA ATPase complex, a complex that mediates the extraction of the PEX5 receptor from peroxisomal membrane (PubMed:12717447, PubMed:12851857, PubMed:16257970, PubMed:16763195, PubMed:16854980, PubMed:21362118)
Peroxisome membrane
Peroxisome biogenesis disorder complementation group 8
A peroxisomal disorder arising from a failure of protein import into the peroxisomal membrane or matrix. The peroxisome biogenesis disorders (PBD group) are genetically heterogeneous with at least 14 distinct genetic groups as concluded from complementation studies. Include disorders are: Zellweger syndrome (ZWS), neonatal adrenoleukodystrophy (NALD), infantile Refsum disease (IRD), and classical rhizomelic chondrodysplasia punctata (RCDP). ZWS, NALD and IRD are distinct from RCDP and constitute a clinical continuum of overlapping phenotypes known as the Zellweger spectrum (PBD-ZSS).
Component of the PEX13-PEX14 docking complex, a translocon channel that specifically mediates the import of peroxisomal cargo proteins bound to PEX5 receptor (PubMed:28765278, PubMed:8858165, PubMed:9653144). The PEX13-PEX14 docking complex forms a large import pore which can be opened to a diameter of about 9 nm (By similarity). Mechanistically, PEX5 receptor along with cargo proteins associates with the PEX14 subunit of the PEX13-PEX14 docking complex in the cytosol, leading to the insertion o
Peroxisome membrane
Peroxisome biogenesis disorder complementation group 13
A peroxisomal disorder arising from a failure of protein import into the peroxisomal membrane or matrix. The peroxisome biogenesis disorders (PBD group) are genetically heterogeneous with at least 14 distinct genetic groups as concluded from complementation studies. Include disorders are: Zellweger syndrome (ZWS), neonatal adrenoleukodystrophy (NALD), infantile Refsum disease (IRD), and classical rhizomelic chondrodysplasia punctata (RCDP). ZWS, NALD and IRD are distinct from RCDP and constitute a clinical continuum of overlapping phenotypes known as the Zellweger spectrum (PBD-ZSS).
This is the major myelin protein from the central nervous system. It plays an important role in the formation or maintenance of the multilamellar structure of myelin
Cell membraneMyelin membrane
Leukodystrophy, hypomyelinating, 1
An X-linked recessive disorder of the central nervous system in which myelin is not formed properly. Clinically characterized by nystagmus, spastic quadriplegia, ataxia, and developmental delay.
Chaperonin implicated in mitochondrial protein import and macromolecular assembly. Together with Hsp10, facilitates the correct folding of imported proteins. May also prevent misfolding and promote the refolding and proper assembly of unfolded polypeptides generated under stress conditions in the mitochondrial matrix (PubMed:11422376, PubMed:1346131). The functional units of these chaperonins consist of heptameric rings of the large subunit Hsp60, which function as a back-to-back double ring. In
Mitochondrion matrix
Spastic paraplegia 13, autosomal dominant
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body.
In neurons, involved in the transport of late endosomes/lysosomes (PubMed:25066864). May be involved in dendrite morphogenesis and maintenance by regulating lysosomal trafficking (PubMed:25066864). May act as a molecular brake for retrograde transport of late endosomes/lysosomes, possibly via its interaction with MAP6 (By similarity). In motoneurons, may mediate the axonal transport of lysosomes and axonal sorting at the initial segment (By similarity). It remains unclear whether TMEM106B affect
Late endosome membraneLysosome membraneCell membrane
Frontotemporal dementia 2
A form of dementia characterized by pathologic finding of frontotemporal lobar degeneration, presenile dementia with behavioral changes, deterioration of cognitive capacities and loss of memory. Gestural apraxia, parkinsonism, visual loss, and visual hallucinations are present in 25 to 40% of patients.
Oxidoreductase that catalyzes the last step in proline biosynthesis, which corresponds to the reduction of pyrroline-5-carboxylate to L-proline using NAD(P)H (PubMed:23024808, PubMed:2722838, PubMed:6894153). At physiologic concentrations, has higher specific activity in the presence of NADH (PubMed:23024808, PubMed:2722838, PubMed:6894153). Involved in cellular response to oxidative stress (PubMed:25865492). In some cell types, such as erythrocytes, its primary function may be the generation of
CytoplasmMitochondrion
Leukodystrophy, hypomyelinating, 10
An autosomal recessive neurologic disorder characterized by postnatal microcephaly, severely delayed psychomotor development, hypomyelination, and reduced cerebral white-matter volume.
Forms a receptor signaling complex with TYROBP which mediates signaling and cell activation following ligand binding (PubMed:10799849). Acts as a receptor for amyloid-beta protein 42, a cleavage product of the amyloid-beta precursor protein APP, and mediates its uptake and degradation by microglia (PubMed:27477018, PubMed:29518356). Binding to amyloid-beta 42 mediates microglial activation, proliferation, migration, apoptosis and expression of pro-inflammatory cytokines, such as IL6R and CCL3, a
Cell membraneSecreted
Polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy 2
An autosomal recessive disease characterized by presenile frontal dementia with leukoencephalopathy and basal ganglia calcification. In most cases the disorder first manifests in early adulthood as pain and swelling in ankles and feet, followed by bone fractures. Neurologic symptoms manifest in the fourth decade of life as a frontal lobe syndrome with loss of judgment, euphoria, and disinhibition. Progressive decline in other cognitive domains begins to develop at about the same time. The disorder culminates in a profound dementia and death by age 50 years.
Voltage-gated and osmosensitive chloride channel. Forms a homodimeric channel where each subunit has its own ion conduction pathway. Conducts double-barreled currents controlled by two types of gates, two fast glutamate gates that control each subunit independently and a slow common gate that opens and shuts off both subunits simultaneously. Displays inward rectification currents activated upon membrane hyperpolarization and extracellular hypotonicity (PubMed:16155254, PubMed:17567819, PubMed:19
Cell membraneBasolateral cell membraneCell projection, dendritic spine membraneCell projection, axon
Epilepsy, idiopathic generalized 11
A disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain.
Forms part of a macromolecular complex that catalyzes the attachment of specific amino acids to cognate tRNAs during protein synthesis (PubMed:25288775). Modulates the secretion of AIMP1 and may be involved in generation of the inflammatory cytokine EMAP2 from AIMP1 (PubMed:17443684)
CytoplasmCytoplasm, cytosol
Leukodystrophy, hypomyelinating, 9
An autosomal recessive neurodegenerative disorder characterized by delayed psychomotor development, severe spasticity, nystagmus, and ataxia associated with diffuse hypomyelination apparent on brain MRI.
Catalytic core component of RNA polymerase III (Pol III), a DNA-dependent RNA polymerase which synthesizes small non-coding RNAs using the four ribonucleoside triphosphates as substrates. Synthesizes 5S rRNA, snRNAs, tRNAs and miRNAs from at least 500 distinct genomic loci (PubMed:19609254, PubMed:19631370, PubMed:20413673, PubMed:33335104, PubMed:33558764, PubMed:33558766, PubMed:34675218, PubMed:35637192, PubMed:9331371). Pol III-mediated transcription cycle proceeds through transcription init
NucleusCytoplasm, cytosol
Leukodystrophy, hypomyelinating, 7, with or without oligodontia and/or hypogonadotropic hypogonadism
An autosomal recessive neurodegenerative disorder characterized by childhood onset of progressive motor decline manifest as spasticity, ataxia, tremor, and cerebellar signs, as well as mild cognitive regression. Other features may include hypodontia or oligodontia and hypogonadotropic hypogonadism. There is considerable inter- and intrafamilial variability.
Zinc ion transporter that mediates zinc efflux and plays a crucial role in intracellular zinc homeostasis (PubMed:25130899, PubMed:31697912, PubMed:36204728). Binds the divalent cations Zn(2+), Ni(2+), and to a minor extent Cu(2+) (By similarity). Is a functional modulator of P2X purinoceptors, including P2RX1, P2RX3, P2RX4 and P2RX7 (PubMed:32492420). Plays a role in central nervous system development and is required for myelination, and survival and proliferation of oligodendrocytes (PubMed:35
Cytoplasmic vesicle, secretory vesicle, synaptic vesicle membraneEarly endosome membraneLate endosome membraneLysosome membraneCell membrane
Leukodystrophy, hypomyelinating, 25
A form of hypomyelinating leukodystrophy, a group of heterogeneous disorders characterized by persistent deficit of myelin observed on brain imaging. HLD25 is an autosomal dominant form with onset in early infancy and characterized by nystagmus, hypotonia, and delayed global development. Most patients show gradual clinical improvement over time with resolution of the nystagmus in early childhood. Many achieve developmental milestones and may have normal cognition, although some affected individuals may have persistent neurologic deficits. Brain imaging shows hypomyelination that may also improve with time.
E3 ubiquitin-protein ligase that promotes the ubiquitination and proteasomal degradation of SIN3B (By similarity). Independently of its E3 ligase activity, acts as a CTNNB1 stabilizer through USP7-mediated deubiquitination of CTNNB1 promoting Wnt signaling (PubMed:25266658, PubMed:33964137). Plays a critical role in the regulation of nuclear lamina (PubMed:33964137)
CytoplasmNucleus
Leukodystrophy, hypomyelinating, 23, with ataxia, deafness, liver dysfunction, and dilated cardiomyopathy
An autosomal recessive neurodegenerative disorder with systemic manifestations. Affected individuals show delayed motor development and ataxic gait in early childhood that progresses to spastic paraplegia with loss of ambulation in the first decades of life. Additional features include progressive sensorineural hearing loss, hepatic dysfunction, and dilated cardiomyopathy. Death occurs in the first or second decades. Brain imaging shows hypomyelination, diffuse white matter abnormalities, and thin corpus callosum.
Ubiquitin-like modifier which can be covalently attached via an isopeptide bond to lysine residues of substrate proteins as a monomer or a lysine-linked polymer (PubMed:15071506, PubMed:20018847, PubMed:27653677, PubMed:29868776, PubMed:30626644, PubMed:38377992, PubMed:38383785). The so-called ufmylation, requires the UFM1-activating E1 enzyme UBA5, the UFM1-conjugating E2 enzyme UFC1, and the UFM1-ligase E3 enzyme UFL1 (PubMed:15071506, PubMed:20018847, PubMed:27653677, PubMed:29868776). Ufmyl
NucleusCytoplasm
Leukodystrophy, hypomyelinating, 14
An autosomal recessive, severe disorder characterized by atrophy of the basal ganglia and cerebellum, hypomyelination, severe developmental delay, typically without intentional movements and language development, and microcephaly. Almost all patients exhibit spasticity, extrapyramidal movement abnormalities, and severe drug-resistant epilepsy. Disease onset is early in infancy, and most patients die in the first years of life.
Hydrolyzes the galactose ester bonds of glycolipids such as galactosylceramide and galactosylsphingosine (PubMed:8281145, PubMed:8399327). Enzyme with very low activity responsible for the lysosomal catabolism of galactosylceramide, a major lipid in myelin, kidney and epithelial cells of small intestine and colon (PubMed:8281145, PubMed:8399327)
Lysosome
Krabbe disease
An autosomal recessive disorder characterized by insufficient catabolism of several galactolipids that are important for normal myelin production. Four clinical forms are recognized. The infantile form accounts for 90% of cases. It manifests before six months of age with irritability, spasticity, arrest of motor and mental development, and bouts of temperature elevation without infection. This is followed by myoclonic jerks of arms and legs, oposthotonus, hypertonic fits, and mental regression, which progresses to a severe decerebrate condition with no voluntary movements and death from respiratory infections or cerebral hyperpyrexia before 2 years of age. Cases with later onset present with unexplained blindness, weakness and sensorimotor peripheral neuropathy, mental deterioration and death.
E3 ubiquitin-protein ligase component of a retrotranslocation channel required for peroxisome organization by mediating export of the PEX5 receptor from peroxisomes to the cytosol, thereby promoting PEX5 recycling (PubMed:24662292). The retrotranslocation channel is composed of PEX2, PEX10 and PEX12; each subunit contributing transmembrane segments that coassemble into an open channel that specifically allows the passage of PEX5 through the peroxisomal membrane (By similarity). PEX10 also regula
Peroxisome membrane
Peroxisome biogenesis disorder complementation group 7
A peroxisomal disorder arising from a failure of protein import into the peroxisomal membrane or matrix. The peroxisome biogenesis disorders (PBD group) are genetically heterogeneous with at least 14 distinct genetic groups as concluded from complementation studies. Include disorders are: Zellweger syndrome (ZWS), neonatal adrenoleukodystrophy (NALD), infantile Refsum disease (IRD), and classical rhizomelic chondrodysplasia punctata (RCDP). ZWS, NALD and IRD are distinct from RCDP and constitute a clinical continuum of overlapping phenotypes known as the Zellweger spectrum (PBD-ZSS).
Component of the PEX13-PEX14 docking complex, a translocon channel that specifically mediates the import of peroxisomal cargo proteins bound to PEX5 receptor (PubMed:24235149, PubMed:28765278, PubMed:9653144). The PEX13-PEX14 docking complex forms a large import pore which can be opened to a diameter of about 9 nm (By similarity). Mechanistically, PEX5 receptor along with cargo proteins associates with the PEX14 subunit of the PEX13-PEX14 docking complex in the cytosol, leading to the insertion
Peroxisome membrane
Peroxisome biogenesis disorder complementation group K
A peroxisomal disorder arising from a failure of protein import into the peroxisomal membrane or matrix. The peroxisome biogenesis disorders (PBD group) are genetically heterogeneous with at least 14 distinct genetic groups as concluded from complementation studies. Include disorders are: Zellweger syndrome (ZWS), neonatal adrenoleukodystrophy (NALD), infantile Refsum disease (IRD), and classical rhizomelic chondrodysplasia punctata (RCDP). ZWS, NALD and IRD are distinct from RCDP and constitute a clinical continuum of overlapping phenotypes known as the Zellweger spectrum (PBD-ZSS).
Involved in the maturation of mitochondrial 4Fe-4S proteins functioning late in the iron-sulfur cluster assembly pathway. May be involved in the binding of an intermediate of Fe/S cluster assembly
Mitochondrion
Multiple mitochondrial dysfunctions syndrome 4
A severe disorder of systemic energy metabolism, resulting in weakness, respiratory failure, lack of neurologic development, lactic acidosis, hyperglycinemia and early death.
Ribonuclease that plays an essential role in innate immune response by recognizing and degrading RNAs from microbial pathogens that are subsequently sensed by TLR8 (PubMed:31778653). Cleaves preferentially single-stranded RNA molecules between purine and uridine residues, which critically contributes to the supply of catabolic uridine and the generation of purine-2',3'-cyclophosphate-terminated oligoribonucleotides (PubMed:31778653, PubMed:38697119). In turn, RNase T2 degradation products promot
SecretedLysosome lumenEndoplasmic reticulum lumenMitochondrion intermembrane space
Leukoencephalopathy, cystic, without megalencephaly
An infantile-onset syndrome of cerebral leukoencephalopathy. Affected newborns develop microcephaly and neurologic abnormalities including psychomotor impairment, seizures and sensorineural hearing impairment. The brain shows multifocal white matter lesions, anterior temporal lobe subcortical cysts, pericystic abnormal myelination, ventriculomegaly and intracranial calcifications.
Receptor that mediates peroxisomal import of proteins containing a C-terminal PTS1-type tripeptide peroxisomal targeting signal (SKL-type) (PubMed:11101887, PubMed:11336669, PubMed:12456682, PubMed:16314507, PubMed:17157249, PubMed:17428317, PubMed:21976670, PubMed:26344566, PubMed:7706321, PubMed:7719337, PubMed:7790377). Binds to cargo proteins containing a PTS1 peroxisomal targeting signal in the cytosol, and translocates them into the peroxisome matrix by passing through the PEX13-PEX14 dock
Cytoplasm, cytosolPeroxisome matrix
Peroxisome biogenesis disorder 2A
A fatal peroxisome biogenesis disorder belonging to the Zellweger disease spectrum and characterized clinically by severe neurologic dysfunction with profound psychomotor retardation, severe hypotonia and neonatal seizures, craniofacial abnormalities, liver dysfunction, and biochemically by the absence of peroxisomes. Additional features include cardiovascular and skeletal defects, renal cysts, ocular abnormalities, and hearing impairment. Most severely affected individuals with the classic form of the disease (classic Zellweger syndrome) die within the first year of life.
Contributes to tetrahydrofolate metabolism. Helps regulate carbon flow through the folate-dependent one-carbon metabolic network that supplies carbon for the biosynthesis of purines, thymidine and amino acids. Catalyzes the irreversible conversion of 5-formyltetrahydrofolate (5-FTHF) to yield 5,10-methenyltetrahydrofolate
Cytoplasm
Neurodevelopmental disorder with microcephaly, epilepsy, and hypomyelination
An autosomal recessive neurodevelopmental disorder with onset at birth or in early infancy, and characterized by microcephaly, short stature, severe global developmental delay, progressive spasticity, and epilepsy. Brain imaging shows delayed myelination, hypomyelination, enlarged ventricles, and cerebellar atrophy.
Catalyzes the attachment of alanine to tRNA(Ala) in a two-step reaction: alanine is first activated by ATP to form Ala-AMP and then transferred to the acceptor end of tRNA(Ala). Also edits incorrectly charged tRNA(Ala) via its editing domain (PubMed:21549344). In presence of high levels of lactate, also acts as a protein lactyltransferase that mediates lactylation of lysine residues in target proteins, such as CGAS (PubMed:39322678). Acts as an inhibitor of cGAS/STING signaling by catalyzing lac
Mitochondrion
Combined oxidative phosphorylation deficiency 8
A mitochondrial disease characterized by a lethal infantile hypertrophic cardiomyopathy, generalized muscle dysfunction and some neurologic involvement. The liver is not affected.
Catalytic core component of RNA polymerase III (Pol III), a DNA-dependent RNA polymerase which synthesizes small non-coding RNAs using the four ribonucleoside triphosphates as substrates. Synthesizes 5S rRNA, snRNAs, tRNAs and miRNAs from at least 500 distinct genomic loci (PubMed:20413673, PubMed:33558766). Pol III-mediated transcription cycle proceeds through transcription initiation, transcription elongation and transcription termination stages. During transcription initiation, Pol III is rec
NucleusCytoplasm, cytosol
Leukodystrophy, hypomyelinating, 8, with or without oligodontia and/or hypogonadotropic hypogonadism
An autosomal recessive neurodegenerative disorder characterized by early childhood onset of cerebellar ataxia and mild intellectual disabilities associated with diffuse hypomyelination apparent on brain MRI. Variable features include oligodontia and/or hypogonadotropic hypogonadism.
One gap junction consists of a cluster of closely packed pairs of transmembrane channels, the connexons, through which materials of low MW diffuse from one cell to a neighboring cell. May play a role in myelination in central and peripheral nervous systems
Cell membraneCell junction, gap junction
Leukodystrophy, hypomyelinating, 2
An autosomal recessive hypomyelinating leukodystrophy with symptoms of Pelizaeus-Merzbacher disease. Clinically characterized by nystagmus, impaired motor development, ataxia, choreoathetotic movements, dysarthria, and progressive spasticity.
Plays a role in vesicle-mediated protein trafficking to lysosomal compartments including the endocytic membrane transport and autophagic pathways. Believed to act as a core component of the putative HOPS and CORVET endosomal tethering complexes which are proposed to be involved in the Rab5-to-Rab7 endosome conversion probably implicating MON1A/B, and via binding SNAREs and SNARE complexes to mediate tethering and docking events during SNARE-mediated membrane fusion. The HOPS complex is proposed
EndosomeLate endosome membraneLysosome membraneEarly endosomeCytoplasmic vesicleCytoplasmic vesicle, autophagosomeCytoplasmic vesicle, clathrin-coated vesicle
Leukodystrophy, hypomyelinating, 12
An autosomal recessive neurologic disorder characterized by developmental delay, spasticity, truncal hypotonia, acquired microcephaly, intellectual disability with variable seizure disorder, accompanied by thin corpus callosum, paucity of white matter and delayed myelination.
Catalyzes the attachment of alanine to tRNA(Ala) in a two-step reaction: alanine is first activated by ATP to form Ala-AMP and then transferred to the acceptor end of tRNA(Ala) (PubMed:27622773, PubMed:27911835, PubMed:28493438, PubMed:33909043). Also edits incorrectly charged tRNA(Ala) via its editing domain (PubMed:27622773, PubMed:27911835, PubMed:28493438, PubMed:29273753). In presence of high levels of lactate, also acts as a protein lactyltransferase that mediates lactylation of lysine res
CytoplasmNucleus
Charcot-Marie-Tooth disease, axonal, type 2N
An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy.
Catalyzes the reversible conversion of ribose-5-phosphate to ribulose 5-phosphate and participates in the first step of the non-oxidative branch of the pentose phosphate pathway
Ribose 5-phosphate isomerase deficiency
An autosomal recessive inborn error of polyols metabolism characterized by highly elevated level of ribitol and arabitol in brain and body fluids. Clinical features include leukoencephalopathy, psychomotor retardation from early life, neurologic regression, and a mild sensorimotor neuropathy.
Required for peroxisome membrane biogenesis. May play a role in early stages of peroxisome assembly. Can recruit other peroxisomal proteins, such as PEX3 and PMP34, to de novo peroxisomes derived from the endoplasmic reticulum (ER). May function as receptor for PEX3
Peroxisome membrane
Peroxisome biogenesis disorder complementation group 9
A peroxisomal disorder arising from a failure of protein import into the peroxisomal membrane or matrix. The peroxisome biogenesis disorders (PBD group) are genetically heterogeneous with at least 14 distinct genetic groups as concluded from complementation studies. Include disorders are: Zellweger syndrome (ZWS), neonatal adrenoleukodystrophy (NALD), infantile Refsum disease (IRD), and classical rhizomelic chondrodysplasia punctata (RCDP). ZWS, NALD and IRD are distinct from RCDP and constitute a clinical continuum of overlapping phenotypes known as the Zellweger spectrum (PBD-ZSS).
Component of the PEX1-PEX6 AAA ATPase complex, a protein dislocase complex that mediates the ATP-dependent extraction of the PEX5 receptor from peroxisomal membranes, an essential step for PEX5 recycling (PubMed:16314507, PubMed:16854980, PubMed:21362118, PubMed:29884772). Specifically recognizes PEX5 monoubiquitinated at 'Cys-11', and pulls it out of the peroxisome lumen through the PEX2-PEX10-PEX12 retrotranslocation channel (PubMed:29884772). Extraction by the PEX1-PEX6 AAA ATPase complex is
Cytoplasm, cytosolPeroxisome membraneCell projection, cilium, photoreceptor outer segment
Peroxisome biogenesis disorder complementation group 4
A peroxisomal disorder arising from a failure of protein import into the peroxisomal membrane or matrix. The peroxisome biogenesis disorders (PBD group) are genetically heterogeneous with at least 14 distinct genetic groups as concluded from complementation studies. Include disorders are: Zellweger syndrome (ZWS), neonatal adrenoleukodystrophy (NALD), infantile Refsum disease (IRD), and classical rhizomelic chondrodysplasia punctata (RCDP). ZWS, NALD and IRD are distinct from RCDP and constitute a clinical continuum of overlapping phenotypes known as the Zellweger spectrum (PBD-ZSS).
Component of a retrotranslocation channel required for peroxisome organization by mediating export of the PEX5 receptor from peroxisomes to the cytosol, thereby promoting PEX5 recycling (PubMed:24662292, PubMed:9354782, PubMed:9632816). The retrotranslocation channel is composed of PEX2, PEX10 and PEX12; each subunit contributing transmembrane segments that coassemble into an open channel that specifically allows the passage of PEX5 through the peroxisomal membrane (By similarity). PEX12 also re
Peroxisome membrane
Peroxisome biogenesis disorder complementation group 3
A peroxisomal disorder arising from a failure of protein import into the peroxisomal membrane or matrix. The peroxisome biogenesis disorders (PBD group) are genetically heterogeneous with at least 14 distinct genetic groups as concluded from complementation studies. Include disorders are: Zellweger syndrome (ZWS), neonatal adrenoleukodystrophy (NALD), infantile Refsum disease (IRD), and classical rhizomelic chondrodysplasia punctata (RCDP). ZWS, NALD and IRD are distinct from RCDP and constitute a clinical continuum of overlapping phenotypes known as the Zellweger spectrum (PBD-ZSS).
Component of the PEX1-PEX6 AAA ATPase complex, a protein dislocase complex that mediates the ATP-dependent extraction of the PEX5 receptor from peroxisomal membranes, an essential step for PEX5 recycling (PubMed:11439091, PubMed:16314507, PubMed:16854980, PubMed:21362118, PubMed:29884772). Specifically recognizes PEX5 monoubiquitinated at 'Cys-11', and pulls it out of the peroxisome lumen through the PEX2-PEX10-PEX12 retrotranslocation channel (PubMed:29884772). Extraction by the PEX1-PEX6 AAA A
Cytoplasm, cytosolPeroxisome membrane
Peroxisome biogenesis disorder complementation group 1
A peroxisomal disorder arising from a failure of protein import into the peroxisomal membrane or matrix. The peroxisome biogenesis disorders (PBD group) are genetically heterogeneous with at least 14 distinct genetic groups as concluded from complementation studies. Include disorders are: Zellweger syndrome (ZWS), neonatal adrenoleukodystrophy (NALD), infantile Refsum disease (IRD), and classical rhizomelic chondrodysplasia punctata (RCDP). ZWS, NALD and IRD are distinct from RCDP and constitute a clinical continuum of overlapping phenotypes known as the Zellweger spectrum (PBD-ZSS).
Involved in regulating cell motility and cell-matrix interactions. May inhibit cell growth through suppression of cell proliferation (PubMed:15885354, PubMed:15917256). In glia, associates and targets CLCN2 at astrocytic processes and myelinated fiber tracts where it may regulate transcellular chloride flux involved in neuron excitability (PubMed:22405205)
CytoplasmCell membrane
Megalencephalic leukoencephalopathy with subcortical cysts 2A
A neurodegenerative disorder characterized by infantile-onset macrocephaly and later onset of motor deterioration, with ataxia and spasticity, seizures, and cognitive decline of variable severity. The brain appears swollen on magnetic resonance imaging with white-matter abnormalities and subcortical cysts, in all stages of the disease.
Tyrosine-protein kinase that acts as a cell-surface receptor for CSF1 and IL34 and plays an essential role in the regulation of survival, proliferation and differentiation of hematopoietic precursor cells, especially mononuclear phagocytes, such as macrophages and monocytes. Promotes the release of pro-inflammatory chemokines in response to IL34 and CSF1, and thereby plays an important role in innate immunity and in inflammatory processes. Plays an important role in the regulation of osteoclast
Cell membrane
Acts as a component of the translation initiation factor 2B (eIF2B) complex, which catalyzes the exchange of GDP for GTP on the eukaryotic initiation factor 2 (eIF2) complex gamma subunit (PubMed:25858979, PubMed:27023709, PubMed:31048492). Its guanine nucleotide exchange factor activity is repressed when bound to eIF2 complex phosphorylated on the alpha subunit, thereby limiting the amount of methionyl-initiator methionine tRNA available to the ribosome and consequently global translation is re
Cytoplasm, cytosol
Leukoencephalopathy with vanishing white matter 3
An autosomal recessive brain disease characterized by neurological features including progressive cerebellar ataxia, spasticity, and cognitive deficits. Brain imaging shows abnormal white matter that vanishes over time and is replaced by cerebrospinal fluid. Disease severity ranges from fatal infantile forms to adult forms without neurological deterioration. The disease is progressive with, in most individuals, additional episodes of rapid deterioration following febrile infections or minor head trauma. Death may occurs after a variable period after disease onset, usually following an episode of fever and coma. A subset of affected females with milder forms of the disease who survive to adolescence exhibit ovarian dysfunction. This variant of the disorder is called ovarioleukodystrophy.
Acts as a component of the translation initiation factor 2B (eIF2B) complex, which catalyzes the exchange of GDP for GTP on eukaryotic initiation factor 2 (eIF2) gamma subunit (PubMed:25858979, PubMed:27023709, PubMed:31048492). Its guanine nucleotide exchange factor activity is repressed when bound to eIF2 complex phosphorylated on the alpha subunit, thereby limiting the amount of methionyl-initiator methionine tRNA available to the ribosome and consequently global translation is repressed (Pub
Cytoplasm, cytosol
Leukoencephalopathy with vanishing white matter 2
An autosomal recessive brain disease characterized by neurological features including progressive cerebellar ataxia, spasticity, and cognitive deficits. Brain imaging shows abnormal white matter that vanishes over time and is replaced by cerebrospinal fluid. Disease severity ranges from fatal infantile forms to adult forms without neurological deterioration. The disease is progressive with, in most individuals, additional episodes of rapid deterioration following febrile infections or minor head trauma. Death may occurs after a variable period after disease onset, usually following an episode of fever and coma. A subset of affected females with milder forms of the disease who survive to adolescence exhibit ovarian dysfunction. This variant of the disorder is called ovarioleukodystrophy.
Acts as a component of the translation initiation factor 2B (eIF2B) complex, which catalyzes the exchange of GDP for GTP on eukaryotic initiation factor 2 (eIF2) gamma subunit (PubMed:25858979, PubMed:27023709, PubMed:31048492). Its guanine nucleotide exchange factor activity is repressed when bound to eIF2 complex phosphorylated on the alpha subunit, thereby limiting the amount of methionyl-initiator methionine tRNA available to the ribosome and consequently global translation is repressed (Pub
Cytoplasm, cytosol
Leukoencephalopathy with vanishing white matter 1
An autosomal recessive brain disease characterized by neurological features including progressive cerebellar ataxia, spasticity, and cognitive deficits. Brain imaging shows abnormal white matter that vanishes over time and is replaced by cerebrospinal fluid. Disease severity ranges from fatal infantile forms to adult forms without neurological deterioration. The disease is progressive with, in most individuals, additional episodes of rapid deterioration following febrile infections or minor head trauma. Death may occurs after a variable period after disease onset, usually following an episode of fever and coma. A subset of affected females with milder forms of the disease who survive to adolescence exhibit ovarian dysfunction. This variant of the disorder is called ovarioleukodystrophy.
Probably functions as a 3'-phosphoadenylyl sulfate:adenosine 3',5'-bisphosphate antiporter at the Golgi membranes. Mediates the transport from the cytosol into the lumen of the Golgi of 3'-phosphoadenylyl sulfate/adenosine 3'-phospho 5'-phosphosulfate (PAPS), a universal sulfuryl donor for sulfation events that take place in that compartment
Golgi apparatus membrane
Leukodystrophy, hypomyelinating, 26, with chondrodysplasia
A form of hypomyelinating leukodystrophy, a group of heterogeneous disorders characterized by persistent deficit of myelin observed on brain imaging. HLD26 is an autosomal recessive form characterized by severe psychomotor delay, limited or absent speech, abnormal development of brain white matter, corpus callosum hypoplasia, and cerebral atrophy. Other features include pre- and postnatal growth retardation, chondrodysplasia, and early-onset scoliosis.
Medicamentos e terapias
Mecanismo: Bile acid receptor FXR agonist
Mecanismo: ARSA exogenous gene
Mecanismo: Peroxisome proliferator-activated receptor gamma agonist
Mecanismo: ABCD1 exogenous gene
Mecanismo: Peroxisome proliferator-activated receptor gamma agonist
Mecanismo: Tyrosine-protein kinase JAK2 inhibitor
Mecanismo: HMG-CoA reductase inhibitor
Mecanismo: Voltage-gated N-type calcium channel alpha-1B subunit blocker
Mecanismo: Glucocorticoid receptor agonist
Variantes genéticas (ClinVar)
185 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 2,898 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
92 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Leucodistrofia
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
11 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
87 ensaios clínicos encontrados, 16 ativos.
Publicações mais relevantes
Mostrando amostra de 200 publicações de um total de 1.939
Progressive gait and motor deficits in a rat model of Alexander disease.
Alexander disease is a leukodystrophy caused by gain-of-function mutations in the gene for Glial Fibrillary Acidic Protein (GFAP) which result in accumulation and aggregation of GFAP protein, astrocyte dysfunction, and ultimately developmental delay, failure to thrive, and intellectual and motor impairment. A Gfap+/R237H rat model, designed to mimic the common R239H human variant, meets normal milestones during early postnatal development, but declines dramatically as the rats mature. At severe stages of disease, Gfap+/R237H rats exhibit cognitive and motor deficits and increased mortality. Here we provide a more detailed analysis of the Gfap+/R237H rat with respect to onset of motor impairments and increasing loss of function. We show that Gfap+/R237H rats develop abnormal open field activity as they mature but stabilize with age, and that motor deficits are apparent as early as 4 weeks of age, as demonstrated by poor rotarod performance. We use automated gait analysis to further characterize subtle differences at this early age and demonstrate the progression and persistence of impairment at late stages of disease. In addition, we find evidence for changes in cerebellar size, suggesting a potential neuroanatomical correlate to the observed deficits. The rat model provides a novel system in which to investigate aspects of impaired motor function and central nervous system pathology that are directly relevant to the human disease.
Natural History of Clinical Phenotypes and Their Biochemical Correlates in Adult X-Linked Adrenoleukodystrophy.
X-linked adrenoleukodystrophy (X-ALD) is a rare monogenic disorder characterized by marked variability in clinical presentation, age at onset, and disease progression. A deeper understanding of its natural history and the relationship between biochemical markers and clinical phenotypes is essential for improving disease monitoring, patient counseling, and optimizing clinical trial design. In particular, the predictive value of very long-chain fatty acids (VLCFA) for clinical phenotypes has recently garnered increased attention. In this longitudinal, mixed prospective/retrospective, single-center study, we analyzed clinical and biochemical data from 364 patients with X-ALD (255 males). Parameters included clinical scores (EDSS, AACS), age at symptom onset, and disease manifestations, which were correlated with individual VLCFA levels. Patients with adrenal insufficiency (AI) exhibited significantly elevated VLCFA concentrations. Higher C26:0 levels were associated with faster progression (measured by EDSS); however, effect sizes were small and inter-individual variability considerable. Although initial symptom severity was comparable between sexes, males presented earlier and progressed faster. Among patients seen in early clinical stages (EDSS ≤ 4.5), disease progression rates were higher (males: 0.34 ± 0.77; females: 0.11 ± 0.11) than in those presenting at more advanced stages (EDSS > 4.5; males: 0.23 ± 0.33; females: 0.09 ± 0.10). We provide comprehensive data on the prevalence of disease manifestations and the natural course of X-ALD in a large adult cohort. The observed association between elevated VLCFA levels and adrenal insufficiency should be considered in future clinical monitoring and trial design. However, due to the small effect sizes and variability, VLCFA levels offer limited prognostic utility for individual patients.
A Depolarizing Leak in Sodium Bicarbonate Cotransporter NBCe1 Causes Brain Edema.
SLC4A4 encodes electrogenic sodium bicarbonate cotransporter NBCe1, prominently expressed in kidney and brain. Recessive loss-of-function variants in SLC4A4 cause proximal renal tubular acidosis, no brain edema. In the brain, NBCe1 is expressed by astrocytes, where it regulates pH and mediates astrocyte volume changes. Here we describe a novel dominant variant in SLC4A4 in patients with brain edema and investigate how it affects NBCe1 function. Genetic studies identified a novel gene variant in three unrelated pediatric patients with the same MRI pattern of cerebral subcortical white matter signal abnormality and swelling, and medulla lesions. Immunohistochemical and electrophysiological experiments were performed to determine the localization of the transporter in the brain and the functional consequence of the patient variant. The same heterozygous variant in SLC4A4 was found in all three patients and one parent. The children displayed infantile-onset progressive macrocephaly, motor and cognitive impairment, autism, epilepsy, and recurrent episodes of increased intracranial pressure. Bicarbonate treatment of two patients led to clinical and MRI improvement. Immunohistochemistry revealed that brain NBCe1 is mainly present in astrocytes, more in cortex than white matter. Functional experiments revealed impaired transporter activity of mutant NBCe1 due to reduced membrane expression and a prominent depolarizing ion leak. The most likely pathomechanism of this novel SLC4A4-related disease is that a depolarizing leak in NBCe1 disrupts astrocyte pH regulation, promoting swelling and impairing volume control. These findings uncover a previously unrecognized mechanism of genetic brain edema and establish NBCe1 as a critical modulator of astrocyte homeostasis.
Alexander Disease: A Literature Review for Clinicians.
Alexander disease (ALEXD; MIM 203450) is a rare leukodystrophy caused by dominant mutations in the GFAP (Glial Fibrillary Acidic Protein) gene, which encodes a key structural protein of astrocytes. First described in 1949, ALEXD is now recognized as a clinically heterogeneous disorder with a broad phenotypic spectrum spanning neonatal, infantile, juvenile, and adult-onset forms. Clinical manifestations vary according to age at onset and disease subtype, ranging from early developmental impairment and progressive neurologic decline to later-onset presentations dominated by bulbar dysfunction, ataxia, and spinal cord involvement. Diagnosis can be challenging because of phenotypic overlap with other neurologic conditions; however, characteristic magnetic resonance imaging patterns combined with molecular confirmation of GFAP mutations are central to diagnosis. To date, more than 100 GFAP mutations have been reported, although robust genotype-phenotype correlations remain elusive. Pathogenic GFAP variants lead to astrocyte dysfunction through protein aggregation, oxidative stress, and cytoskeletal disorganization, resulting in Rosenthal fiber formation. Elevated GFAP levels in cerebrospinal fluid have emerged as a potential biomarker, though their clinical utility remains under investigation. Current management is supportive, but emerging gene-targeted approaches, including antisense oligonucleotides such as zilganersen and AAV-mediated gene silencing strategies, offer promising therapeutic prospects. This review provides an updated overview of the clinical, radiologic, genetic, and molecular features of ALEXD, emphasizing its spectrum of phenotypes across age groups and ongoing efforts toward improved diagnosis and targeted treatment.
Brain morphometry and cognition in late-onset glutaric aciduria type 1: scoping review and novel insights from a case report.
Glutaric Aciduria type I (GA1) is a rare autosomal recessive organic aciduria, with typical early-onset presentation, characterized by severe movement disorders with damage to the basal ganglia following an acute encephalopathic crisis. Late-onset (LO), milder forms have rarely been described. In LO patients, severe brain damage is frequently reported, including frontotemporal hypoplasia, brain atrophy, subependymal nodules and leukodystrophy. Cognitive impairment is sometimes described, often without formal psychometric assessment. This scoping review aims to synthesize the neuroradiological and neuropsychological features of all LO GA1 patients reported in the literature to date, divided in undiagnosed LO cases detected through selective screening and LO cases with onset of symptoms after 6 years of age. The following databases were used: PubMed, Embase and Medline. The search strategy abides by the PRISMA-ScR guidelines. Out of the 53 LO patients reviewed, extensive quantitative neuropsychological testing was reported in seven cases, while no brain morphometric analysis was performed. CASE REPORT. A novel case of a GA1 34-year-old Chinese woman identified through the neonatal screening of her healthy baby is described. Brain morphometric analysis showed diffused reduced volumes and cortical thinning, involving fronto-temporal areas and, to a lesser extent, the parieto-occipital regions. The neuropsychological assessment highlighted mild difficulties in verbal executive functions (inferential thinking) and phonological short-term memory. The present review and case report suggest that integrating neuroanatomical and neuropsychological investigations into clinical practice may allow a more refined characterization of GA1 patients, contributing to unveil the complex ethio-pathogenic mechanisms underlying the disease and monitor patients over time. The online version contains supplementary material available at 10.1007/s10072-026-08886-9.
Publicações recentes
LAMB1-related adult-onset leukoencephalopathy presenting with cognitive decline: diagnostic workup and review of the literature.
🥉 Relato de casoFinding comfort in complexity: The role of palliative care in children with leukodystrophy.
🥉 Relato de casoNovel NDUFV1 variant in progressive cavitating leukodystrophy with microcephaly: a case report.
🥉 Relato de casoGeneration of an isogenic human induced pluripotent stem cell line harbouring a CLDN11 mutation associated with hypomyelinating leukodystrophy.
Adult-Onset Leukodystrophies Mimicking Multiple Sclerosis.
📚 EuropePMC2.000 artigos no totalmostrando 197
Progressive gait and motor deficits in a rat model of Alexander disease.
Behavioural brain researchPretreatment blood NfL indicates response to cellular therapies in cerebral adrenoleukodystrophy.
Communications medicineNatural History of Clinical Phenotypes and Their Biochemical Correlates in Adult X-Linked Adrenoleukodystrophy.
Journal of inherited metabolic diseaseA Depolarizing Leak in Sodium Bicarbonate Cotransporter NBCe1 Causes Brain Edema.
Annals of clinical and translational neurologyAlexander Disease: A Literature Review for Clinicians.
Journal of child neurologyA Wolf in Metabolic Clothing: Familial HLH Mimicking a Neurometabolic Disorder.
Journal of child neurologyMultiparametric MRI analysis of clinical outcome after hematopoietic stem cell transplantation in juvenile Metachromatic Leukodystrophy.
AJNR. American journal of neuroradiologyGenetic screening of EIF2B genes reveals mutation spectrum and predicted prevalence of vanishing white matter disease in Chinese population.
Clinica chimica acta; international journal of clinical chemistryDesign of a Pediatric Low Motor Function Item Battery in leukodystrophies.
Molecular genetics and metabolismBrain morphometry and cognition in late-onset glutaric aciduria type 1: scoping review and novel insights from a case report.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyJuvenile Metachromatic Leukodystrophy in a Seven-Year-Old Child With a Familial History: A Case Report Suggesting Saposin B Deficiency.
CureusExpanding the genotypic spectrum of combined oxidative phosphorylation deficiency 54.
NeurogeneticsMetachromatic leukodystrophy (MLD) in France: the views of family caregivers on the diagnosis of the disease, its daily burden on their child, and the whole family.
Archives de pediatrie : organe officiel de la Societe francaise de pediatrieRecessive PPTC7 deficiency triggers excessive mitophagy to cause a severe inborn error of metabolism with hypomyelinating leukodystrophy.
Research squareMutant PLP1 impairs COPII vesicle formation via ER calcium depletion in Pelizaeus-Merzbacher disease.
Neurobiology of diseaseSphingolipid-neutralizing molecular therapy reduces psychosine cytotoxicity in Krabbe disease.
iScienceRNA-based discovery and correction of splicing defects caused by POLR3A missense mutations.
Molecular therapy. Nucleic acidsMyelin oligodendrocyte glycoprotein antibody disease (MOGAD) with leukodystrophy-like presentation in an adult.
Acta neurologica BelgicaEIF2AK2-related hypomyelinating leukoencephalopathy presenting with congenital bilateral vocal cord paralysis and a spinal cord lesion.
NeurogeneticsInfantile-Onset Vanishing White Matter Disease in an Azerbaijani Infant With a Homozygous EIF2B5 p.(Arg195His) Variant.
CureusWhite Matter Matters: A Magnetic Resonance Imaging Study with Clinical Correlates in Primary Brain Calcification.
Movement disorders : official journal of the Movement Disorder SocietyCompound heterozygosity of a novel missense variant and exonic deletion in hypomyelinating leukodystrophy 15.
NeurogeneticsLongitudinal MRI-based changes in intracranial volume and skull thickness observed in both metachromatic leukodystrophy and multiple sclerosis.
NeuroImage. ClinicalPOLR3A-related syndrome complicated with cerebral abscesses: a case report and literature review.
Frontiers in geneticsA canine PLP1 missense variant differentiates oligodendrocyte maturation in connatal and classical Pelizaeus-Merzbacher disease.
Proceedings of the National Academy of Sciences of the United States of AmericaAdult-onset vanishing white matter disease caused by the EIF2B5 c.185A>T (p.Asp62Val) variant.
Frontiers in geneticsConsensus-based follow-up and treatment registry for GNAO1-associated disorder.
Developmental medicine and child neurologyVery late-onset Krabbe disease with concomitant dementia: case description and a critical review of the literature.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyNovel VARS1 variants define new clinical and molecular subtypes of a rare neurodevelopmental syndrome.
Biochimica et biophysica acta. Molecular basis of diseaseA novel patient-Centered approach to clinical trial readiness in rare diseases: Application in Aicardi-Goutières Syndrome (AGS).
Molecular genetics and metabolismThe Grey Zone Project: Risk-Based Classification of ABCD1 Variants in X-Linked Adrenoleukodystrophy.
Journal of inherited metabolic diseaseBrachial plexopathy in juvenile-onset Krabbe disease: A rare case.
Skeletal radiologyDefaceQA - automated quality assessment of brain MRI defacing software.
BMC medical imagingAstrocyte-specific deletion of LRRC8A causes neurological dysfunction but not chronic white matter edema.
Neurobiology of diseaseUse of Brain MRI in Cerebral Adrenoleukodystrophy: International Recommendations for Screening, Monitoring, and Research.
NeurologyConsensus Guidelines for Imaging in Adrenoleukodystrophy.
NeurologyCSF1R-related leukoencephalopathy presenting with early apathy, hypoactivity, and cognitive flattening: a case report of a diagnostic challenge.
Frontiers in human neuroscienceGenetic Disorders That Confer Risk for the Development of Psychosis and White Matter Hyperintensities on T2-weighted Imaging: A Practical Guide for Psychiatrists.
Innovations in clinical neuroscienceExpanding the Genotype Spectrum- A Novel POLR3B Variant in 4H Leukodystrophy.
Annals of Indian Academy of Neurology[Hypogonadotropic hypogonadism due to pathogenic variants in the POLR3B gene].
Problemy endokrinologiiLeukodystrophy-like phenotype in early-onset neuropsychiatric systemic lupus erythematosus: Case series and systematic review of the literature.
Revue neurologiqueCSF1R mutations in an Italian population of early-onset dementia: a case series.
Journal of neurologyLeukoencephalopathy with Brainstem and Spinal Cord Involvement and Lactate Elevation Presenting Primarily with Exercise Intolerance: A Case Report.
Annals of clinical and laboratory scienceProsaposin in CNS health and disease, metabolic stress and exercise adaptation.
Journal of molecular medicine (Berlin, Germany)Is it a secondary cause of vasculitis or a mimic? A case of retinal vasculopathy with cerebral leukoencephalopathy.
Modern rheumatology case reportsExploring targeted therapy in retinal vasculopathy with cerebral leukoencephalopathy: a case report and review of literature.
Frontiers in immunologyLong-term neurological outcome after hematopoietic stem cell transplant in juvenile Krabbe disease.
Journal of neurologyA case report of multiple system atrophy-mimics: Importance of comprehensive evaluation in suspected familial cases.
MedicineSpatially resolved lipids in a mouse brain model of globoid cell leukodystrophy via IR-MALDESI MSI and parallel reaction monitoring MSI.
Analytical and bioanalytical chemistryNeurocognitive Outcome After Pediatric Traumatic Brain Injury: Patient Subgroups With Diverging Outcome.
Pediatric neurologyTherapeutic suppression of Tubb4a rescues H-ABC leukodystrophy.
Molecular therapy : the journal of the American Society of Gene TherapyConfirmation of biallelic VPS11 variants as a cause of complex dystonic syndrome.
Clinical parkinsonism & related disordersSexual and developmental variability of spike-wave discharges in the taiep rat: A model of H-ABC leukodystrophy.
Epilepsy researchCongenital Inborn Errors of Metabolism: Clinical and Imaging Pearls.
Radiographics : a review publication of the Radiological Society of North America, IncGallbladder mucinous carcinoma in a child with metachromatic leukodystrophy, case report and literature review.
BMC pediatricsThe Impact of RNA Polymerase III-Related Leukodystrophy on Nonaffected Family Members: A Qualitative Study.
Pediatric neurologyA case report of integrating Chinese and Western medicine: A new era in the treatment of late-onset Krabbe disease.
TransplantationCharacterization of Clinical Phenotype to Glial Fibrillary Acidic Protein Concentrations in Alexander Disease.
Annals of clinical and translational neurologyEvolution of the lipidome uncovers early changes in adrenoleukodystrophy human cortical and spinal organoids.
iScienceMitochondrial DNA Maintenance Defects: Clinical, Imaging, and Genetic Spectrum of Four Patients from a Single Tertiary Care Centre.
Annals of Indian Academy of NeurologyCaregiver-reported disease burden in Krabbe disease: evaluating outcomes of hematopoietic stem cell transplantation.
Orphanet journal of rare diseasesHemiconvulsion-hemiplegia-epilepsy syndrome in a child with an underlying hypomyelinating leukodystrophy: a previously unreported association.
Pediatric radiologyOligodendrocyte mechanotransduction channel TMEM63A regulates myelin sheath geometry.
NeuronMegalencephalic leukoencephalopathy with subcortical cysts: a multicenter Italian experience.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyMild and late onset forms of type I 3-methylglutaconic aciduria presenting as isolated cerebellar ataxia without leukodystrophy: case reports and phenotype expansion.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyThe Era of Gene Therapy, Newborn Screening, and Improved Management in the Leukodystrophies: A Shifting Framework With Altered Expectations.
Pediatric neurologyDe novo variants in KDM2A cause a syndromic neurodevelopmental disorder.
American journal of human geneticsAbnormal Splicing of GALC Transcripts Underlies Unusual Cases of Krabbe Disease.
BiomedicinesA Truncating Variant in the ERCC6 Gene With Three Different Phenotypes: Significant Effects of Modifier Genes.
Genetics researchIntegrated stress response inhibition prolongs the lifespan of a Pelizaeus-Merzbacher disease mouse model by increasing oligodendrocyte survival.
Nature communicationsAssociation Between Rapid Progression, Early Mortality, and Imaging in Neonatal-Onset Alexander Disease.
NeuropediatricsDurable Global Correction of CNS and PNS and Lifespan Rescue in Murine Globoid Cell Leukodystrophy via AAV9-Mediated Monotherapy.
CellsImpaired docking and recycling of synaptic vesicles in inherited lysosomal sphingolipidoses.
Cell communication and signaling : CCSNew multiplex LC-MS/MS method for lipid biomarker analysis of inherited neurodegenerative metabolic diseases.
Journal of lipid researchJuvenile metachromatic leukodystrophy caused by a rare genetic mutation.
NeurogeneticsNavigating the Uncommon: "Juvenile-Onset Huntington Disease".
Journal of child neurologyGenetic and Clinical Characteristics of Chinese Adult Patients With Krabbe Disease.
CNS neuroscience & therapeuticsDEGS1-Related Hypomyelinating Leukodystrophy: Four Individuals From Same Family and Review of Literature.
Neurology. GeneticsCase Report: A pharmacist-led precision therapy framework for managing invasive fungal infection in CSF1R-Related leukoencephalopathy post Allo-HSCT.
Frontiers in pharmacologyChorea in Hereditary Leukodystrophies - Overview of Two Cases.
Tremor and other hyperkinetic movements (New York, N.Y.)Gene therapy for Krabbe disease: evidence from mouse and canine models.
GeneLiver Transplantation and Other Hepatically Directed Therapies Do Not Change the Biochemical Phenotype nor Halt Progression of Leukodystrophy due to Biallelic HMBS Variants: A Case Report.
JIMD reportsCRISPR-mediated genomic repair of ARSA mutations in metachromatic leukodystrophy: a transformative step toward precision neuromodulation.
Annals of medicine and surgery (2012)Astrocytes differentiated from patient iPSCs model the rare leukodystrophy MLC and uncover disease-linked maturation defects and Kir4.1 channel dysfunction.
Neurobiology of diseaseAdult-onset Familial TUBB4A-related Leukodystrophy Caused by c.286G>A (p.Gly96Arg) in a Korean Family: A Case Report.
Journal of movement disordersFrom Neonatal Encephalopathy to Adult Survival: Revisiting the Natural History of D-Bifunctional Protein Deficiency in a Multicentre International Case Series.
Journal of inherited metabolic diseaseA sulfatide-centered ultra-high-resolution magnetic resonance MALDI imaging benchmark dataset for MS1-based lipid annotation tools.
GigaScienceSpatial and temporal brain biodistribution of neuropathogenic sphingolipids of Krabbe disease.
Journal of lipid researchGenotype-phenotype correlations of GFAP variants in type I Alexander disease subtypes.
Molecular genetics and metabolismSaposin B Deficiency With Neurologic and Hepatobiliary Involvement: Two Patients Expanding the Clinical Spectrum.
Journal of child neurologyAltered Mechanical Properties of Astrocytes Lacking MLC1: Implications for the Leukodystrophy MLC.
GliaOne year follow up in siblings with TREX1-associated retinal vasculopathy with cerebral leukodystrophy.
American journal of ophthalmology case reportsNeurological manifestations and clinical outcomes in pediatric Alexander disease: single-center cohort and identification of novel GFAP variants.
Brain & developmentA scoping review of stem cell models of leukodystrophies: advances in understanding pathophysiological mechanisms.
NPJ genomic medicineRegulation of the orphan G-protein-coupled receptor GPRC5B by MLC1 and the cell adhesion molecule GlialCAM in megalencephalic leukoencephalopathy.
The Journal of biological chemistryPsychological Framing of Illness: Early Family Trauma and Diagnostic Delay in Adult-Onset Metachromatic Leukodystrophy.
Case reports in psychiatryFacing the challenge of effective dosing, safety, and timing of intrathecal gene therapy for neurological disorders.
EBioMedicineAgrawal Disease (Megalencephalic Leukoencephalopathy with Subcortical Cysts): A Rare Neurological Case Report.
Annals of African medicineAn automated classification of brain white matter inherited disorders (Leukodystrophy) using MRI image features.
Biomedical physics & engineering express[Epileptic encephalopathy associated with a mutation in the KCNT1 gene].
Zhurnal nevrologii i psikhiatrii imeni S.S. KorsakovaNewborn Screening for Metachromatic Leukodystrophy: A Systematic Literature Review.
International journal of neonatal screeningGeneralized tonic-clonic seizures as the initial symptom of late-onset Krabbe disease: a Case Report.
Frontiers in behavioral neuroscienceCeramide levels predict clinical severity in adult-onset Krabbe disease independent of extensive white matter hyperintensities.
Neurobiology of diseaseAnalyzing accessibility and suitability of online Krabbe disease resources.
Journal of community genetics"Case report": Whole-exome sequencing reveals compound heterozygous variants in the EIF2B5 gene in a familial case of vanishing white matter.
Frontiers in geneticsChronic interferon-alpha overexpression induces white matter damage and neurovascular abnormalities in a mouse model of Aicardi-Goutières syndrome.
Experimental neurologyConsensus-Based Expert Recommendations for Diagnosis and Clinical Management of Vanishing White Matter.
NeurologyCognitive and intellectual functioning in leukodystrophy patients: a systematic review.
Orphanet journal of rare diseasesA novel homozygous loss-of-function NOTCH3 variant in a Moroccan patient: expanding the spectrum beyond CADASIL.
NeurogeneticsPropionic Acidemia: Gray Matter Disease Meets Subcortical Leukodystrophy.
Journal of inherited metabolic diseaseRisk of Seizures and Epilepsy in the Leukodystrophies.
Pediatric neurologyComments on: "Biallelic ELOVL1 Variants Are Linked to Hypomyelinating Leukodystrophy, Movement Disorder, and Ichthyosis".
Movement disorders : official journal of the Movement Disorder SocietyMitochondrial DNA Depletion Syndrome 1 (MTDPS1)-A Novel Cause of Premature Ovarian Insufficiency.
Clinical geneticsDifferentiating Incidental From Pathologic Brain MRI Findings in Asymptomatic Boys With X-Linked Adrenoleukodystrophy: A Multicenter Study.
NeurologyImpact of HLD6-associated TUBB4A mutant proteins on cell morphogenesis.
BMC research notesEndogenous Repair in Vanishing White Matter.
Annals of neurologyDeveloping a National Network for Leukodystrophy Research and Care in Canada: The CARELeuko Initiative.
Neurology. GeneticsPelizaeus-Merzbacher disease in children: A case report.
Radiology case reportsAn Automated Analysis Tool for Diffusion Tensor Imaging-Based Quantitative MRI in X-Linked Adrenoleukodystrophy.
Journal of inherited metabolic diseaseMethodological and Procedural Considerations for Developing Decision Analytic Models to Assess the Health Economic Impacts of Newborn Bloodspot Screening: A Systematic Methodological Review.
International journal of neonatal screeningCritical Functional Domains in Pediatric Onset TUBB4A-Related Leukodystrophy: A Clinical and Caregiver's Perspective.
Pediatric neurologyHypomyelination With Congenital Cataract: A Rare Genetic Leukodystrophy.
Cureus[A case of adult-onset Krabbe disease diagnosed by galactocerebrosidase gene mutations, presenting with an atypical phenotype].
Rinsho shinkeigaku = Clinical neurologyExacerbation of absence seizures by central prolactin in female taiep rats: An animal model of epilepsy-prone leukodystrophy.
Epilepsy & behavior : E&BHypogonadism in adult males with adrenoleukodystrophy.
Annales d'endocrinologieDescription of the Hamburg Alexander Leukodystrophy Cohort-Insights into Practical Classification and the Care Situation.
Journal of clinical medicineBeyond Krabbe disease, the intriguing connection of galactocerebrosidase (GALC) with nervous system illness: A novel risk factor?
NeuroscienceEncapsulated cells as an enzyme replacement therapy for metachromatic leukodystrophy.
Journal of controlled release : official journal of the Controlled Release SocietyMyelin-water imaging and multi-shell diffusion-weighted imaging in adults with adrenoleukodystrophy.
Brain communicationsAssessment of Potential Side Effects Related To RAB27A Gene Therapy in Stem Cells.
Stem cell reviews and reportsMutations in the Key Autophagy Tethering Factor EPG5 Link Neurodevelopmental and Neurodegenerative Disorders Including Early-Onset Parkinsonism.
Annals of neurologyDiagnosing and Managing Pelizaeus-Merzbacher Disease: A Pediatric Struggle.
Clinical case reportsTwo Japanese families with adult-onset leukoencephalopathy caused by pathogenic variants in CST3.
Journal of the neurological sciencesImproving quality of life in rare diseases using disease-specific, multidisciplinary online interventions on the example of rare X-linked adrenoleukodystrophy: a randomized-controlled trial.
Therapeutic advances in neurological disordersLymphomatosis cerebri presenting with rapidly progressive parkinsonism and Holmes tremor: a case report.
BMC neurologyPediatric Influenza-Associated Encephalopathy and Acute Necrotizing Encephalopathy - United States, 2024-25 Influenza Season.
MMWR. Morbidity and mortality weekly reportChimeric enzymes enhance treatment potential for globoid cell leukodystrophy through hematopoietic stem cell gene therapy.
Molecular therapy : the journal of the American Society of Gene TherapyNeurocognitive Network Organization in Children with Traumatic Brain Injury.
Journal of neurotraumaPOLR3B-Related Hypomyelinating Leukodystrophy Type 8 (4H Syndrome): A Case Series of Two Siblings.
CureusOligodendrocyte-targeted adeno-associated virus gene therapy for Canavan disease in children: a phase 1/2 trial.
Nature medicineMolecular Characterization of the GALC Mutation Thr112Ala Causing Krabbe Disease.
International journal of molecular sciencesClinical Practice Guidelines for the Diagnosis, Management, and Surveillance of LMNB1-Related Autosomal Dominant Leukodystrophy.
Neurology. GeneticsHypomyelinating leukodystrophy: From molecular mechanisms to clinical advances.
Brain & developmentPOLR3 gene and protein expression dynamics in 4H leukodystrophy using iPSC-derived neuronal lineages.
Stem cell researchDominant MLC-causing mutations alter hepaCAM subcellular localization and protein interactome in astrocytes of the developing mouse cortex.
bioRxiv : the preprint server for biologyTwo Novel Mutations in the PSAP Gene Causing a Metachromatic Leukodystrophy (MLD)-like Phenotype and a Review of the Literature.
Neurology IndiaPrevalence of lysosomal storage disease (LSD) in Malaysia.
The Malaysian journal of pathologyClinically Important Endpoints in Individuals With Leukodystrophy: A Multisite Study.
Annals of the Child Neurology SocietyLoss of dihydroceramide desaturase drives neurodegeneration by disrupting endoplasmic reticulum and lipid droplet homeostasis in glial cells.
eLifeThe impact of vanishing white matter on unaffected family members.
Orphanet journal of rare diseasesUnravelling neurodegeneration with cerebral calcifications: Krabbe disease masquerading as Aicardi-Goutieres syndrome.
BMJ case reportsThe impact of leukodystrophies on parents' lives.
Journal of pediatric psychologyNeuroimaging Spectrum of GM1 Gangliosidosis with Description of Novel Imaging Signs.
AJNR. American journal of neuroradiologySHQ1-related hypomyelinating leukodystrophy: A case report with imaging features and a homozygous variant.
Radiology case reportsStem cell and gene therapies for leukodystrophies.
Molecular therapy. Methods & clinical developmentUncommon Allies: Van der Knaap Syndrome and Focal Segmental Glomerulosclerosis.
The Journal of the Association of Physicians of IndiaPathogenic ZNF319 variant disrupts nuclear localization and transcriptional regulation to cause a novel form of autosomal recessive leukodystrophy.
Journal of human geneticsApplication of the Gross Motor Function Measure in children with conditions other than cerebral palsy: A systematic review.
Developmental medicine and child neurologyClinical Characterization of a Multicenter International Cohort of Patients With Aicardi-Goutières Syndrome Homozygous for the RNASEH2B:p.Ala177Thr Variant: Early Clinical Markers of Disease Severity.
Pediatric neurologyKIF1C-related disorders: spastic ataxia or hypomyelinating leukodystrophy? A paradigm of classification ambiguity.
NeurogeneticsBLOC1S1 variants cause lysosomal and autophagic defects resulting in a hypomyelinating leukodystrophy with epileptic encephalopathy.
medRxiv : the preprint server for health sciencesEvidence Regarding Metachromatic Leukodystrophy Newborn Screening.
PediatricsA focus on the normal-appearing white and gray matter within the multiple sclerosis brain: a link to smoldering progression.
Acta neuropathologicaGene therapy for lysosomal storage diseases.
Brain & developmentClinical and Intestinal Ultrasound Findings in Mitochondrial Neurogastrointestinal Encephalomyopathy:Report of One Case.
Zhongguo yi xue ke xue yuan xue bao. Acta Academiae Medicinae SinicaeA monoallelic 8q24.3-duplication involving a single protein encoding TSNARE1 gene may be linked to a new leukodystrophy.
NeurogeneticsARSA Variants Associated With Cognitive Decline and Long-Term Preservation of Motor Function in Metachromatic Leukodystrophy.
Journal of inherited metabolic diseaseRecurrent Hemorrhagic Stroke and Microcephaly in a Newborn with Aicardi-Goutières Syndrome Caused by a Homozygous Intronic RNASEH2B Variant.
Annals of clinical and laboratory scienceInherited white matter disorders in Japan: focusing on demyelinating leukodystrophy.
Brain & developmentOutcome of two siblings with late-onset Krabbe disease following allogeneic hematopoietic stem cell transplantation: And review of literature.
Molecular genetics and metabolism reportsCochlear implantation in Childhood Ataxia with Central nervous system Hypomyelination Syndrome.
Indian journal of otolaryngology and head and neck surgery : official publication of the Association of Otolaryngologists of IndiaCost-Effectiveness of Newborn Screening for X-Linked Adrenoleukodystrophy in the Netherlands: A Health-Economic Modelling Study.
International journal of neonatal screeningInflammation and Immunomodulation in Cerebral X-linked Adrenoleukodystrophy: Review of Pathology and Interventions.
Journal of child neurologyTMEM63A, associated with hypomyelinating leukodystrophies, is an evolutionarily conserved regulator of myelination.
Proceedings of the National Academy of Sciences of the United States of AmericaAmelioration of Inflammation and Metabolic Blockage in GALC Deficient Mice After Enzyme Replacement Therapy via Extracellular Vesicles.
International journal of nanomedicineComprehensive genotype-phenotype analysis in POLR3-related disorders.
HGG advancesChronic integrated stress response causes dysregulated cholesterol synthesis in white matter disease.
JCI insightThe neurological pathology of peroxisomal ACBD5 deficiency - lessons from patients and mouse models.
Frontiers in molecular neuroscienceUnderstanding the molecular basis of the mutation in the RNA polymerase III subunit Rpc10 (R41W)- associated with hypomyelinating leukodystrophy in the yeast homolog Rpc11.
Biochemical and biophysical research communicationsA Novel Pathogenic Variant Identified in HIKESHI-Related Hypomyelinating Leukodystrophy Disrupts Heat Shock Response in iPSCs.
International journal of molecular sciencesHematopoietic Stem Cell Transplantation in an International Cohort of Colony Stimulating Factor-1 Receptor (CSF1R)-Related Disorder.
Movement disorders : official journal of the Movement Disorder SocietyLenmeldy (atidarsagene autotemcel) for individuals with early metachromatic leukodystrophy (MLD): A therapeutics bulletin of the American College of Medical Genetics and Genomics (ACMG).
Genetics in medicine openAdult-Onset Alexander Disease: A Case Report and Literature Review of Glu207 Alterations.
CureusHypomyelination Leukodystrophy Type 11 (HLD11) Presenting with Diabetes: A Case Report and Literature Review.
Sage open pediatricsScreening for Life: Perspectives From Adult Metabolic Specialists on Newborn Screening for Inherited Metabolic Diseases.
Journal of inherited metabolic diseaseClinical Vigilance in Rare Disease Management: Atypical Features Lead to Discovery of Concurrent X-linked Adrenoleukodystrophy and Cystic Fibrosis.
Journal of child neurologySelf-Assembly of Accumulated Sphingolipids into Cytotoxic Fibrils in Globoid Cell Leukodystrophy and Their Inhibition by Small Molecules In Vitro.
ACS nanoThe first report of a successful birth by preimplantation genetic testing for leukodystrophy induced by IBA57 gene.
Taiwanese journal of obstetrics & gynecologyOverview of genetic variants in a cohort of Iranian patients with leukodystrophy.
Scientific reportsUncommon Non-MS Demyelinating Disorders of the Central Nervous System.
Current neurology and neuroscience reportsBiallelic ELOVL1 Variants Are Linked to Hypomyelinating Leukodystrophy, Movement Disorder, and Ichthyosis.
Movement disorders : official journal of the Movement Disorder SocietyInvestigating the Cellular Effects of GALC Dosing in Enzyme Replacement Therapy for Krabbe Disease Supports the Role of Nanomedicine.
Advanced biologyMetachromatic Leukodystrophy: New Therapy Advancements and Emerging Research Directions.
NeurologyMicroglia loss triggers glial stress and white matter damage in human leukodystrophy.
Nature immunologyPeripheral Neuropathy as an Early Marker in Newborn-Screened Krabbe Disease: The Value of Pre-Confirmatory Neurophysiological Testing.
Journal of the peripheral nervous system : JPNSMyelin oligodendrocyte glycoprotein-antibody disease (MOGAD) with leukodystrophy-like presentation.
Practical neurologyMolecular pathologies and therapies for Pelizaeus-Merzbacher disease.
Brain & developmentAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Leucodistrofia.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Leucodistrofia
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Progressive gait and motor deficits in a rat model of Alexander disease.
- Natural History of Clinical Phenotypes and Their Biochemical Correlates in Adult X-Linked Adrenoleukodystrophy.
- A Depolarizing Leak in Sodium Bicarbonate Cotransporter NBCe1 Causes Brain Edema.
- Alexander Disease: A Literature Review for Clinicians.
- Brain morphometry and cognition in late-onset glutaric aciduria type 1: scoping review and novel insights from a case report.Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology· 2026· PMID 41779049mais citado
- LAMB1-related adult-onset leukoencephalopathy presenting with cognitive decline: diagnostic workup and review of the literature.
- Finding comfort in complexity: The role of palliative care in children with leukodystrophy.
- Novel NDUFV1 variant in progressive cavitating leukodystrophy with microcephaly: a case report.
- Generation of an isogenic human induced pluripotent stem cell line harbouring a CLDN11 mutation associated with hypomyelinating leukodystrophy.
- Adult-Onset Leukodystrophies Mimicking Multiple Sclerosis.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:68356(Orphanet)
- MONDO:0019046(MONDO)
- GARD:6895(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q1821559(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar