Doença rara que afeta o transporte e absorção de aminoácidos, manifestando-se com problemas oculares (retinopatia, oftalmoplegia), auditivos, ceratocone e alterações metabólicas como hiperinsulinemia.
Introdução
O que você precisa saber de cara
Visão geral
A doença do transporte de aminoácidos é uma condição rara que afeta a forma como o corpo absorve e transporta os aminoácidos, que são os blocos de construção das proteínas. Isso pode levar a uma variedade de problemas de saúde, pois os aminoácidos são essenciais para o funcionamento de muitos órgãos e sistemas. A condição é classificada como um erro inato do metabolismo e está listada na Classificação Internacional de Doenças (CID-10) sob o código E72.0.[1][2]
Sinais e sintomas
Os sinais e sintomas da doença do transporte de aminoácidos podem variar amplamente de pessoa para pessoa. Eles podem incluir problemas renais, como tubulopatia proximal (um tipo de disfunção dos túbulos renais), hematúria microscópica (sangue na urina visível apenas ao microscópio) e acidose metabólica (acúmulo de ácido no corpo). Também podem ocorrer alterações oculares, como ceratocone (córnea em formato de cone), morfologia anormal da córnea, dor ocular, oftalmoplegia (paralisia dos músculos oculares) e anormalidades na retina, incluindo perda do segmento externo dos fotorreceptores e alterações na pigmentação. Problemas neurológicos como ataxia cerebelar progressiva (falta de coordenação motora), disartria espástica (dificuldade na fala), marcha de base alargada e atrofia cerebelar difusa também podem estar presentes. Outros sintomas relatados incluem megacariocitopenia (baixa contagem de plaquetas), hiperinsulinemia (excesso de insulina no sangue), deficiência auditiva condutiva, retrognatia (mandíbula recuada), anormalidades da tireoide e do metabolismo da vitamina D, além de hipovolemia (volume sanguíneo baixo).[1][3]
Causas genéticas
A doença do transporte de aminoácidos tem causas genéticas complexas. Vários genes já foram associados a essa condição, incluindo PRPH2, CLTRN, PREPL, SCN1A, KCNB1, CELF2, CACNA2D1, PACS2, NECAP1, FBXO28, DNM1 e IMPG1. Cada um desses genes fornece instruções para a produção de proteínas que desempenham papéis importantes em diferentes partes do corpo, como no sistema nervoso, nos rins e nos olhos. Mutações (alterações) nesses genes podem prejudicar o transporte adequado de aminoácidos, levando aos sintomas observados.[1][4]
Diagnóstico
O diagnóstico da doença do transporte de aminoácidos geralmente envolve uma combinação de exames clínicos e laboratoriais. Exames de sangue e urina podem ser solicitados para dosar os níveis de aminoácidos e ácidos orgânicos, ajudando a identificar possíveis erros inatos do metabolismo. O teste do pezinho (triagem neonatal) também pode detectar alguns desses distúrbios precocemente. Para confirmar o diagnóstico genético, o sequenciamento completo do exoma (WES) é um exame disponível que analisa todos os genes de uma pessoa em busca de mutações. Atualmente, existem 336 testes genéticos disponíveis e 785 variantes genéticas registradas no ClinVar, um banco de dados público que reúne informações sobre a relação entre variantes genéticas e doenças.[1][4]
Tratamento e manejo
O tratamento e o manejo da doença do transporte de aminoácidos são focados no controle dos sintomas e na melhora da qualidade de vida. Não há um tratamento único, e o plano de cuidados deve ser individualizado. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para essa condição, incluindo procedimentos como a dosagem de aminoácidos, a dosagem de ácidos orgânicos na urina, o teste de triagem para erros inatos do metabolismo, o sequenciamento completo do exoma (WES), o teste do pezinho (triagem neonatal), o atendimento em reabilitação para doenças raras e a fórmula metabólica para erros inatos do metabolismo (como na fenilcetonúria - PKU). É fundamental que o paciente seja acompanhado por uma equipe multidisciplinar de saúde para monitorar e tratar as diferentes manifestações da doença.[1]
Prognóstico e qualidade de vida
O prognóstico para pessoas com doença do transporte de aminoácidos pode variar significativamente dependendo da gravidade dos sintomas e dos órgãos afetados. O acompanhamento médico regular e o manejo adequado dos sintomas são essenciais para melhorar a qualidade de vida e prevenir complicações. Como se trata de uma condição rara e complexa, o suporte de uma equipe multidisciplinar e o acesso a informações atualizadas são fundamentais para o paciente e sua família.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Doença rara que afeta o transporte e absorção de aminoácidos, manifestando-se com problemas oculares (retinopatia, oftalmoplegia), auditivos, ceratocone e alterações metabólicas como hiperinsulinemia.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A doença do transporte de aminoácidos é uma condição rara que afeta a forma como o corpo absorve e transporta os aminoácidos, que são os blocos de construção das proteínas. Isso pode levar a uma variedade de problemas de saúde, pois os aminoácidos são essenciais para o funcionamento de muitos órgãos e sistemas. A condição é classificada como um erro inato do metabolismo e está listada na Classificação Internacional de Doenças (CID-10) sob o código E72.0.[1][2]
Sinais e sintomas
Os sinais e sintomas da doença do transporte de aminoácidos podem variar amplamente de pessoa para pessoa. Eles podem incluir problemas renais, como tubulopatia proximal (um tipo de disfunção dos túbulos renais), hematúria microscópica (sangue na urina visível apenas ao microscópio) e acidose metabólica (acúmulo de ácido no corpo). Também podem ocorrer alterações oculares, como ceratocone (córnea em formato de cone), morfologia anormal da córnea, dor ocular, oftalmoplegia (paralisia dos músculos oculares) e anormalidades na retina, incluindo perda do segmento externo dos fotorreceptores e alterações na pigmentação. Problemas neurológicos como ataxia cerebelar progressiva (falta de coordenação motora), disartria espástica (dificuldade na fala), marcha de base alargada e atrofia cerebelar difusa também podem estar presentes. Outros sintomas relatados incluem megacariocitopenia (baixa contagem de plaquetas), hiperinsulinemia (excesso de insulina no sangue), deficiência auditiva condutiva, retrognatia (mandíbula recuada), anormalidades da tireoide e do metabolismo da vitamina D, além de hipovolemia (volume sanguíneo baixo).[1][3]
Causas genéticas
A doença do transporte de aminoácidos tem causas genéticas complexas. Vários genes já foram associados a essa condição, incluindo PRPH2, CLTRN, PREPL, SCN1A, KCNB1, CELF2, CACNA2D1, PACS2, NECAP1, FBXO28, DNM1 e IMPG1. Cada um desses genes fornece instruções para a produção de proteínas que desempenham papéis importantes em diferentes partes do corpo, como no sistema nervoso, nos rins e nos olhos. Mutações (alterações) nesses genes podem prejudicar o transporte adequado de aminoácidos, levando aos sintomas observados.[1][4]
Diagnóstico
O diagnóstico da doença do transporte de aminoácidos geralmente envolve uma combinação de exames clínicos e laboratoriais. Exames de sangue e urina podem ser solicitados para dosar os níveis de aminoácidos e ácidos orgânicos, ajudando a identificar possíveis erros inatos do metabolismo. O teste do pezinho (triagem neonatal) também pode detectar alguns desses distúrbios precocemente. Para confirmar o diagnóstico genético, o sequenciamento completo do exoma (WES) é um exame disponível que analisa todos os genes de uma pessoa em busca de mutações. Atualmente, existem 336 testes genéticos disponíveis e 785 variantes genéticas registradas no ClinVar, um banco de dados público que reúne informações sobre a relação entre variantes genéticas e doenças.[1][4]
Tratamento e manejo
O tratamento e o manejo da doença do transporte de aminoácidos são focados no controle dos sintomas e na melhora da qualidade de vida. Não há um tratamento único, e o plano de cuidados deve ser individualizado. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para essa condição, incluindo procedimentos como a dosagem de aminoácidos, a dosagem de ácidos orgânicos na urina, o teste de triagem para erros inatos do metabolismo, o sequenciamento completo do exoma (WES), o teste do pezinho (triagem neonatal), o atendimento em reabilitação para doenças raras e a fórmula metabólica para erros inatos do metabolismo (como na fenilcetonúria - PKU). É fundamental que o paciente seja acompanhado por uma equipe multidisciplinar de saúde para monitorar e tratar as diferentes manifestações da doença.[1]
Prognóstico e qualidade de vida
O prognóstico para pessoas com doença do transporte de aminoácidos pode variar significativamente dependendo da gravidade dos sintomas e dos órgãos afetados. O acompanhamento médico regular e o manejo adequado dos sintomas são essenciais para melhorar a qualidade de vida e prevenir complicações. Como se trata de uma condição rara e complexa, o suporte de uma equipe multidisciplinar e o acesso a informações atualizadas são fundamentais para o paciente e sua família.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 253 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 639 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
83 genes identificados com associação a esta condição.
Essential for retina photoreceptor outer segment disk morphogenesis, may also play a role with ROM1 in the maintenance of outer segment disk structure (By similarity). Required for the maintenance of retinal outer nuclear layer thickness (By similarity). Required for the correct development and organization of the photoreceptor inner segment (By similarity)
MembraneCell projection, cilium, photoreceptor outer segmentPhotoreceptor inner segment
Retinitis pigmentosa 7
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Plays an important role in amino acid transport by acting as binding partner of amino acid transporters SLC6A18 and SLC6A19, regulating their trafficking on the cell surface and their amino acid transporter activity (By similarity). May also play a role in trafficking of amino acid transporters SLC3A1 and SLC7A9 to the renal cortical cell membrane (By similarity). Regulator of SNARE complex function (PubMed:16330323). Stimulator of beta cell replication (PubMed:16330323)
Cell membrane
Serine peptidase whose precise substrate specificity remains unclear (PubMed:16143824, PubMed:16385448, PubMed:28726805). Does not cleave peptides after a arginine or lysine residue (PubMed:16143824). Regulates trans-Golgi network morphology and sorting by regulating the membrane binding of the AP-1 complex (PubMed:23321636). May play a role in the regulation of synaptic vesicle exocytosis (PubMed:24610330)
Cytoplasm, cytosolGolgi apparatus, trans-Golgi networkCytoplasm, cytoskeletonGolgi apparatusNucleus
Hypotonia-cystinuria syndrome
Characterized generalized hypotonia at birth, nephrolithiasis, growth hormone deficiency, minor facial dysmorphism, failure to thrive, followed by hyperphagia and rapid weight gain in late childhood.
Pore-forming subunit of Nav1.1, a voltage-gated sodium (Nav) channel that directly mediates the depolarizing phase of action potentials in excitable membranes. Navs, also called VGSCs (voltage-gated sodium channels) or VDSCs (voltage-dependent sodium channels), operate by switching between closed and open conformations depending on the voltage difference across the membrane. In the open conformation they allow Na(+) ions to selectively pass through the pore, along their electrochemical gradient.
Cell membrane
Generalized epilepsy with febrile seizures plus 2
A rare autosomal dominant, familial condition with incomplete penetrance and large intrafamilial variability. Patients display febrile seizures persisting sometimes beyond the age of 6 years and/or a variety of afebrile seizure types. This disease combines febrile seizures, generalized seizures often precipitated by fever at age 6 years or more, and partial seizures, with a variable degree of severity.
Voltage-gated potassium channel that mediates transmembrane potassium transport in excitable membranes, primarily in the brain, but also in the pancreas and cardiovascular system. Contributes to the regulation of the action potential (AP) repolarization, duration and frequency of repetitive AP firing in neurons, muscle cells and endocrine cells and plays a role in homeostatic attenuation of electrical excitability throughout the brain (PubMed:23161216). Plays also a role in the regulation of exo
Cell membranePerikaryonCell projection, axonCell projection, dendriteMembranePostsynaptic cell membraneSynapseSynapse, synaptosomeLateral cell membraneCell membrane, sarcolemma
Developmental and epileptic encephalopathy 26
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE26 patients manifest multiple types of seizures, delayed psychomotor development, poor or absent speech, hypotonia, hypsarrhythmia.
RNA-binding protein implicated in the regulation of several post-transcriptional events. Involved in pre-mRNA alternative splicing, mRNA translation and stability. Mediates exon inclusion and/or exclusion in pre-mRNA that are subject to tissue-specific and developmentally regulated alternative splicing. Specifically activates exon 5 inclusion of TNNT2 in embryonic, but not adult, skeletal muscle. Activates TNNT2 exon 5 inclusion by antagonizing the repressive effect of PTB. Acts both as an activ
NucleusCytoplasm
Developmental and epileptic encephalopathy 97
A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE97 is an autosomal dominant form.
The alpha-2/delta subunit of voltage-dependent calcium channels regulates calcium current density and activation/inactivation kinetics of the calcium channel (PubMed:35293990). Plays an important role in excitation-contraction coupling (By similarity)
MembraneCell membrane
Developmental and epileptic encephalopathy 110
A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE110 is an autosomal recessive form characterized by profound global developmental delay and hypotonia apparent in infancy followed by onset of seizures in the first months or years of life.
Multifunctional sorting protein that controls the endoplasmic reticulum (ER)-mitochondria communication, including the apposition of mitochondria with the ER and ER homeostasis. In addition, in response to apoptotic inducer, translocates BIB to mitochondria, which initiates a sequence of events including the formation of mitochondrial truncated BID, the release of cytochrome c, the activation of caspase-3 thereby causing cell death. May also be involved in ion channel trafficking, directing acid
Endoplasmic reticulumMitochondrion
Developmental and epileptic encephalopathy 66
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE66 is an autosomal dominant form characterized by onset of seizures in first days or weeks of life.
Involved in endocytosis
Cytoplasmic vesicle, clathrin-coated vesicle membraneCell membrane
Developmental and epileptic encephalopathy 21
A severe disease characterized by intractable seizures, profound global developmental delay, and persistent severe axial hypotonia as well as appendicular hypertonia.
Substrate-recognition component of the SCF (SKP1-CUL1-F-box protein)-type E3 ubiquitin ligase complex, promoting ubiquitination and proteasomal degradation of specific target proteins including TOP2A, RAB27A or itself (PubMed:27754753, PubMed:31678254). Regulates topoisomerase IIalpha/TOP2A decatenation activity and plays an important role in maintaining genomic stability (PubMed:27754753). Plays a role in lipid metabolism and inflammation through the ubiquitinated degradation of RAB27A (By simi
Chromosome, centromere, kinetochoreNucleusChromosome
Developmental and epileptic encephalopathy 100
A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE100 is an autosomal dominant, severe form characterized by global developmental delay and onset of variable types of seizures in the first months or years of life.
Catalyzes the hydrolysis of GTP and utilizes this energy to mediate vesicle scission and participates in many forms of endocytosis, such as clathrin-mediated endocytosis or synaptic vesicle endocytosis as well as rapid endocytosis (RE) (PubMed:15703209, PubMed:20428113, PubMed:29668686, PubMed:8101525, PubMed:8910402, PubMed:9362482). Associates to the membrane, through lipid binding, and self-assembles into rings and stacks of interconnected rings through oligomerization to form a helical polym
Cell membraneMembrane, clathrin-coated pitCytoplasmic vesiclePresynapseCytoplasmic vesicle, secretory vesicle, chromaffin granule
Developmental and epileptic encephalopathy 31A
An autosomal dominant epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent.
Chondroitin sulfate-, heparin- and hyaluronan-binding protein (By similarity). May serve to form a basic macromolecular scaffold comprising the insoluble interphotoreceptor matrix (PubMed:9813076)
Cell projection, cilium, photoreceptor outer segmentSecreted, extracellular space, extracellular matrix, interphotoreceptor matrixPhotoreceptor inner segment
Macular dystrophy, vitelliform, 4
A form of macular dystrophy, a retinal disease in which various forms of deposits, pigmentary changes, and atrophic lesions are observed in the macula lutea. Vitelliform macular dystrophies are characterized by yellow, lipofuscin-containing deposits, usually localized at the center of the macula. VMD4 features include late-onset moderate visual impairment, small satellite drusen-like lesions in the foveal area, and preservation of retinal pigment epithelium reflectivity.
Ligand-gated anion channel that allows the movement of anions across cell membranes when activated by calcium (Ca2+) (PubMed:11904445, PubMed:12907679, PubMed:18179881, PubMed:18400985, PubMed:19853238, PubMed:21330666, PubMed:26200502, PubMed:26720466, PubMed:35789156). Allows the movement of chloride and hydrogencarbonate (PubMed:11904445, PubMed:12907679, PubMed:18179881, PubMed:18400985, PubMed:19853238, PubMed:21330666, PubMed:26200502, PubMed:26720466, PubMed:35789156). Found in a partiall
Cell membraneBasolateral cell membrane
Macular dystrophy, vitelliform, 2
An autosomal dominant form of macular degeneration that usually begins in childhood or adolescence. VMD2 is characterized by typical 'egg-yolk' macular lesions due to abnormal accumulation of lipofuscin within and beneath the retinal pigment epithelium cells. Progression of the disease leads to destruction of the retinal pigment epithelium and vision loss.
The BBSome complex is thought to function as a coat complex required for sorting of specific membrane proteins to the primary cilia. The BBSome complex is required for ciliogenesis but is dispensable for centriolar satellite function. This ciliogenic function is mediated in part by the Rab8 GDP/GTP exchange factor, which localizes to the basal body and contacts the BBSome. Rab8(GTP) enters the primary cilium and promotes extension of the ciliary membrane. Firstly the BBSome associates with the c
Cell projection, cilium membraneCytoplasmCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centriolar satellite
Bardet-Biedl syndrome 2
A syndrome characterized by usually severe pigmentary retinopathy, early-onset obesity, polydactyly, hypogenitalism, renal malformation and intellectual disability. Secondary features include diabetes mellitus, hypertension and congenital heart disease. Bardet-Biedl syndrome inheritance is autosomal recessive, but three mutated alleles (two at one locus, and a third at a second locus) may be required for clinical manifestation of some forms of the disease.
Required for the maintenance and formation of cilia. Plays an indirect role in hedgehog (Hh) signaling, cilia being required for all activity of the hedgehog pathway (By similarity)
Cell projection, cilium
Short-rib thoracic dysplasia 10 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Participates in processes of transmission and amplification of the visual signal. cGMP-PDEs are the effector molecules in G-protein-mediated phototransduction in vertebrate rods and cones
Retinitis pigmentosa 57
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Transcription factor that binds and transactivates the sequence 5'-TAATC[CA]-3' which is found upstream of several photoreceptor-specific genes, including the opsin genes. Acts synergistically with other transcription factors, such as NRL, RORB and RAX, to regulate photoreceptor cell-specific gene transcription. Essential for the maintenance of mammalian photoreceptors
Nucleus
Leber congenital amaurosis 7
A severe dystrophy of the retina, typically becoming evident in the first years of life. Visual function is usually poor and often accompanied by nystagmus, sluggish or near-absent pupillary responses, photophobia, high hyperopia and keratoconus.
Ligand-activated transcription factor that enables cells to adapt to changing conditions by sensing compounds from the environment, diet, microbiome and cellular metabolism, and which plays important roles in development, immunity and cancer (PubMed:23275542, PubMed:30373764, PubMed:32818467, PubMed:7961644). Upon ligand binding, translocates into the nucleus, where it heterodimerizes with ARNT and induces transcription by binding to xenobiotic response elements (XRE) (PubMed:23275542, PubMed:30
CytoplasmNucleus
Retinitis pigmentosa 85
A form of retinitis pigmentosa, a retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. RP85 is an autosomal recessive form manifesting as early-onset progressive difficulty to adapt in dim light and gradually decreasing visual acuity in both eyes.
Synaptic vesicle transporter with apparent selectivity for neutral amino acids. The transport is sodium-coupled but chloride-independent, likely driven by the proton electrochemical gradient generated by vacuolar H(+)-ATPase in an overall electrogenic mechanism. May contribute to the synaptic uptake of neurotransmitter precursors in a process coupled in part to vesicle exocytosis
Cytoplasmic vesicle, secretory vesicle, synaptic vesicle membranePostsynapsePresynapse
Intellectual developmental disorder, autosomal recessive 48
A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT48 patients show moderate to severe intellectual disability and additional features including progressive tremor, speech impairment, and sometimes behavioral problems.
Mediates the Na(+)- and Cl(-)-dependent uptake of imino acids such as L-proline, N-methyl-L-proline and pipecolate as well as N-methylated amino acids (PubMed:15632147, PubMed:19033659, PubMed:33428810). Also transports glycine, regulates proline and glycine homeostasis in the brain playing a role in the modulation of NMDAR currents (PubMed:33428810)
Apical cell membrane
Hyperglycinuria
A condition characterized by excess of glycine in the urine. In some cases it is associated with renal colic and renal oxalate stones.
Adapter protein implicated in the regulation of a large spectrum of both general and specialized signaling pathways (PubMed:15696159, PubMed:16511572, PubMed:36732624). Binds to a large number of partners, usually by recognition of a phosphoserine or phosphothreonine motif (PubMed:15696159, PubMed:16511572, PubMed:36732624). Binding generally results in the modulation of the activity of the binding partner (PubMed:16511572). Promotes inactivation of WDR24 component of the GATOR2 complex by bindi
Cytoplasm, cytosolMitochondrion matrix
Developmental and epileptic encephalopathy 56
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE56 is an autosomal dominant condition.
As part of the KICSTOR complex functions in the amino acid-sensing branch of the TORC1 signaling pathway. Recruits, in an amino acid-independent manner, the GATOR1 complex to the lysosomal membranes and allows its interaction with GATOR2 and the RAG GTPases. Functions upstream of the RAG GTPases and is required to negatively regulate mTORC1 signaling in absence of amino acids. In absence of the KICSTOR complex mTORC1 is constitutively localized to the lysosome and activated. The KICSTOR complex
Lysosome membranePeroxisome
Developmental and epileptic encephalopathy 18
A severe autosomal recessive neurologic disorder characterized by lack of psychomotor development apparent from birth, dysmorphic facial features, early onset of refractory seizures, and thick corpus callosum and persistent cavum septum pellucidum on brain imaging.
Voltage-gated potassium channel that mediates transmembrane potassium transport in excitable membranes, primarily in the brain. Contributes to the regulation of the fast action potential repolarization and in sustained high-frequency firing in neurons of the central nervous system. Homotetramer channels mediate delayed-rectifier voltage-dependent potassium currents that activate rapidly at high-threshold voltages and inactivate slowly. Forms tetrameric channels through which potassium ions pass
Cell membraneMembranePerikaryonCell projection, axonCell projection, dendritePostsynaptic cell membranePresynaptic cell membraneSynapse, synaptosomeSynapseApical cell membraneBasolateral cell membrane
Catalytic subunit of the V1 complex of vacuolar(H+)-ATPase (V-ATPase), a multisubunit enzyme composed of a peripheral complex (V1) that hydrolyzes ATP and a membrane integral complex (V0) that translocates protons (PubMed:8463241). V-ATPase is responsible for acidifying and maintaining the pH of intracellular compartments and in some cell types, is targeted to the plasma membrane, where it is responsible for acidifying the extracellular environment (PubMed:32001091). In aerobic conditions, invol
CytoplasmCytoplasm, cytosolCytoplasmic vesicle, secretory vesicleCytoplasmic vesicle, clathrin-coated vesicle membraneLysosome
Cutis laxa, autosomal recessive, 2D
A form of cutis laxa, a disorder characterized by an excessive congenital skin wrinkling, a large fontanelle with delayed closure, a typical facial appearance with downslanting palpebral fissures, and a general connective tissue weakness. Most ARCL2D patients exhibit severe hypotonia as well as cardiovascular and neurologic involvement.
Subunit of non-clathrin- and clathrin-associated adaptor protein complex 3 (AP-3) that plays a role in protein sorting in the late-Golgi/trans-Golgi network (TGN) and/or endosomes. The AP complexes mediate both the recruitment of clathrin to membranes and the recognition of sorting signals within the cytosolic tails of transmembrane cargo molecules. AP-3 appears to be involved in the sorting of a subset of transmembrane proteins targeted to lysosomes and lysosome-related organelles. In concert w
Cytoplasmic vesicle, clathrin-coated vesicle membraneGolgi apparatus
Developmental and epileptic encephalopathy 48
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE48 is an autosomal recessive form characterized by onset of seizures in the first year of life. Affected individuals manifest global developmental delay, intellectual disability, absent speech, and poor, if any, motor development.
Pore-forming subunit of the rod cyclic nucleotide-gated channel. Mediates rod photoresponses at dim light converting transient changes in intracellular cGMP levels into electrical signals. In the dark, cGMP levels are high and keep the channel open enabling a steady inward current carried by Na(+) and Ca(2+) ions that leads to membrane depolarization and neurotransmitter release from synaptic terminals. Upon photon absorption cGMP levels decline leading to channel closure and membrane hyperpolar
Cell membrane
Retinitis pigmentosa 49
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Receptor tyrosine kinase that transduces signals from the extracellular matrix into the cytoplasm by binding to several ligands including LGALS3, TUB, TULP1 or GAS6. Regulates many physiological processes including cell survival, migration, differentiation, and phagocytosis of apoptotic cells (efferocytosis). Ligand binding at the cell surface induces autophosphorylation of MERTK on its intracellular domain that provides docking sites for downstream signaling molecules. Following activation by l
Cell membrane
Retinitis pigmentosa 38
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
The BBSome complex is thought to function as a coat complex required for sorting of specific membrane proteins to the primary cilia. The BBSome complex is required for ciliogenesis but is dispensable for centriolar satellite function. This ciliogenic function is mediated in part by the Rab8 GDP/GTP exchange factor, which localizes to the basal body and contacts the BBSome. Rab8(GTP) enters the primary cilium and promotes extension of the ciliary membrane. Firstly the BBSome associates with the c
Cytoplasm, cytoskeleton, microtubule organizing center, centrosomeCell projection, cilium membraneCytoplasmCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centriolar satelliteCell projection, cilium
Retinitis pigmentosa 51
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Adapter protein that is involved in microtubule polymerization, and spindle function by stabilizing microtubules and anchoring them at centrosomes. Therefore, ensures mitotic progression and genome stability (PubMed:27030108). Acts as a central regulator of microtubule reorganization in apico-basal epithelial differentiation (By similarity). Plays a role during oocyte meiosis by regulating microtubule dynamics (By similarity). Participates in neurite growth by interacting with plexin B3/PLXNB3 a
Cytoplasm, cytoskeleton
Skin creases, congenital symmetric circumferential, 2
An autosomal dominant disease characterized by multiple, symmetric, circumferential rings of folded skin, affecting primarily the limbs. Affected individuals also exhibit intellectual disability, cleft palate, and dysmorphic features.
Acts as a chaperone that facilitates biogenesis and trafficking of functional transporter heteromers to the plasma membrane (By similarity) (PubMed:10588648, PubMed:11318953, PubMed:16609684, PubMed:16825196, PubMed:32494597, PubMed:32817565, PubMed:7686906, PubMed:8486766, PubMed:8663184, PubMed:8663357). Associates with SLC7A9 to form a functional transporter complex that mediates the electrogenic exchange between cationic amino acids and neutral amino acids, with a stoichiometry of 1:1. SLC7A
Cell membraneApical cell membrane
Cystinuria
An autosomal disorder characterized by impaired epithelial cell transport of cystine and dibasic amino acids (lysine, ornithine, and arginine) in the proximal renal tubule and gastrointestinal tract. The impaired renal reabsorption of cystine and its low solubility causes the formation of calculi in the urinary tract, resulting in obstructive uropathy, pyelonephritis, and, rarely, renal failure.
Involved in vision
Cell projection, cilium, photoreceptor outer segmentMembraneEndoplasmic reticulumGolgi apparatus
Retinitis pigmentosa 36
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Essential for the regulation of ciliary length and required for the long-term survival of photoreceptors (By similarity). Phosphorylates FZR1 in a cell cycle-dependent manner. Plays a role in the transcriptional coactivation of AR. Could play an important function in spermatogenesis. May play a role in chromosomal stability in prostate cancer cells
NucleusCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, spindleMidbodyCell projection, cilium, photoreceptor outer segmentPhotoreceptor inner segment
Retinitis pigmentosa 62
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Catalyzes the ATP-dependent unwinding of U4/U6 RNA duplices, an essential step in the assembly of a catalytically active spliceosome (PubMed:35241646). Plays a role in pre-mRNA splicing as a core component of precatalytic, catalytic and postcatalytic spliceosomal complexes (PubMed:28502770, PubMed:28781166, PubMed:29301961, PubMed:29360106, PubMed:29361316, PubMed:30315277, PubMed:30705154, PubMed:30728453). As a component of the minor spliceosome, involved in the splicing of U12-type introns in
Nucleus
Retinitis pigmentosa 33
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
The BBSome complex is thought to function as a coat complex required for sorting of specific membrane proteins to the primary cilia. The BBSome complex is required for ciliogenesis but is dispensable for centriolar satellite function. This ciliogenic function is mediated in part by the Rab8 GDP/GTP exchange factor, which localizes to the basal body and contacts the BBSome. Rab8(GTP) enters the primary cilium and promotes extension of the ciliary membrane. Firstly the BBSome associates with the c
Cell projection, cilium membraneCytoplasmCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centriolar satellite
G-protein coupled receptor for glutamate. Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling activates a phosphatidylinositol-calcium second messenger system. May participate in the central action of glutamate in the CNS, such as long-term potentiation in the hippocampus and long-term depression in the cerebellum (PubMed:24603153, PubMed:28886343, PubMed:7476890).
Cell membranePostsynaptic cell membraneCell projection, dendrite
Spinocerebellar ataxia, autosomal recessive, 13
A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCAR13 is characterized by delayed psychomotor development beginning in infancy. Affected individuals show mild to profound intellectual disability with poor or absent speech as well as gait and stance ataxia and hyperreflexia.
Sodium-dependent, high-affinity amino acid transporter that mediates the uptake of L-glutamate and also L-aspartate and D-aspartate (PubMed:21123949, PubMed:26690923, PubMed:33658209, PubMed:7521911, PubMed:7914198, PubMed:8857541). Can also transport L-cysteine (PubMed:21123949). Functions as a symporter that transports one amino acid molecule together with two or three Na(+) ions and one proton, in parallel with the counter-transport of one K(+) ion (PubMed:26690923, PubMed:33658209, PubMed:75
Cell membraneApical cell membraneSynapse, synaptosomeEarly endosome membraneLate endosome membraneRecycling endosome membrane
Dicarboxylic aminoaciduria
An autosomal recessive disorder characterized by abnormal excretion of urinary glutamate and aspartate, resulting from the incomplete reabsorption of anionic amino acids from the glomerular filtrate in the kidney. It can be associated with intellectual disability.
Acts as a guanine-nucleotide releasing factor (GEF) for RAB8A and RAB37 by promoting the conversion of inactive RAB-GDP to the active form RAB-GTP (PubMed:20631154). GEF activity towards RAB8A may facilitate ciliary trafficking by modulating ciliary intracellular localization of RAB8A (PubMed:20631154). GEF activity towards RAB37 maintains autophagic homeostasis and retinal function (By similarity). Involved in photoreceptor integrity (By similarity). May control cilia formation by regulating ac
Cytoplasm, cytoskeleton, flagellum axonemeGolgi apparatusCell projection, ciliumCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, cilium axoneme
Retinitis pigmentosa 3
An X-linked retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. In RP3, affected males have a severe phenotype, and carrier females show a wide spectrum of clinical features ranging from completely asymptomatic to severe retinitis pigmentosa. Heterozygous women can manifest a form of choroidoretinal degeneration which is distinguished from other types by the absence of visual defects in the presence of a brilliant, scintillating, golden-hued, patchy appearance most striking around the macula, called a tapetal-like retinal reflex.
Involved in ciliogenesis
Cytoplasm, cytoskeleton, cilium basal bodyCell projection, ciliumCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centriole
Retinitis pigmentosa 28
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Has no detectable ceramide-kinase activity. Overexpression of CERKL protects cells from apoptosis in oxidative stress conditions
CytoplasmNucleus, nucleolusGolgi apparatus, trans-Golgi networkEndoplasmic reticulum
Retinitis pigmentosa 26
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Plays a role in pre-mRNA splicing as component of the U4/U6-U5 tri-snRNP complex that is involved in spliceosome assembly, and as component of the precatalytic spliceosome (spliceosome B complex)
NucleusNucleus speckle
Retinitis pigmentosa 18
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Required for normal development of photoreceptor synapses. Required for normal photoreceptor function and for long-term survival of photoreceptor cells. Interacts with cytoskeleton proteins and may play a role in protein transport in photoreceptor cells (By similarity). Binds lipids, especially phosphatidylinositol 3-phosphate, phosphatidylinositol 4-phosphate, phosphatidylinositol 5-phosphate, phosphatidylinositol 3,4-bisphosphate, phosphatidylinositol 4,5-bisphosphate, phosphatidylinositol 3,4
CytoplasmCell membraneSecretedSynapse
Retinitis pigmentosa 14
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Transporter that mediates resorption of neutral amino acids across the apical membrane of renal and intestinal epithelial cells (PubMed:15286787, PubMed:15286788, PubMed:18424768, PubMed:18484095, PubMed:19185582, PubMed:26240152). This uptake is sodium-dependent and chloride-independent (PubMed:15286787, PubMed:15286788, PubMed:19185582). Requires CLTRN in kidney or ACE2 in intestine for cell surface expression and amino acid transporter activity (PubMed:18424768, PubMed:19185582)
Cell membraneApical cell membrane
Hartnup disorder
Autosomal recessive abnormality of renal and gastrointestinal neutral amino acid transport noted for its clinical variability. First described in 1956, HND is characterized by increases in the urinary and intestinal excretion of neutral amino acids. Individuals with typical Hartnup aminoaciduria may be asymptomatic, some develop a photosensitive pellagra-like rash, attacks of cerebellar ataxia and other neurological or psychiatric features. Although the definition of HND was originally based on clinical and biochemical abnormalities, its marked clinical heterogeneity has led to it being known as a disorder with a consistent pathognomonic neutral hyperaminoaciduria.
Voltage-sensitive calcium channels (VSCC) mediate the entry of calcium ions into excitable cells and are also involved in a variety of calcium-dependent processes, including muscle contraction, hormone or neurotransmitter release, gene expression, cell motility, cell division and cell death. The isoform alpha-1A gives rise to P and/or Q-type calcium currents. P/Q-type calcium channels belong to the 'high-voltage activated' (HVA) group and are specifically blocked by the spider omega-agatoxin-IVA
Cell membrane
Spinocerebellar ataxia 6
Spinocerebellar ataxia is a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA6 is an autosomal dominant cerebellar ataxia (ADCA), mainly caused by expansion of a CAG repeat in the coding region of CACNA1A. There seems to be a correlation between the repeat number and earlier onset of the disorder.
High-affinity sodium/citrate cotransporter that mediates the entry of citrate into cells, which is a critical participant of biochemical pathways (PubMed:12445824, PubMed:12826022, PubMed:26324167, PubMed:26384929, PubMed:30054523, PubMed:33597751, PubMed:39622972). May function in various metabolic processes in which citrate has a critical role such as energy production (Krebs cycle), fatty acid synthesis, cholesterol synthesis, glycolysis, and gluconeogenesis (PubMed:12826022). Transports citr
Cell membrane
Developmental and epileptic encephalopathy 25, with amelogenesis imperfecta
An autosomal recessive disease characterized by subclinical seizures appearing in the first days of life, evolving to severe epileptic disease. Affected individuals have profound or severe delayed development with lack of speech, and most patients do not acquire the ability to sit. Additional variable features include axial hypotonia, peripheral hypertonia, and abnormal involuntary movements such as dystonia and choreoathetosis. Dental abnormalities, including delayed eruption, hypodontia, tooth hypoplasia, yellow discoloration, thin enamel, and enamel chipping are observed in most patients.
Hyperpolarization-activated ion channel that are permeable to sodium and potassium ions (PubMed:15351778, PubMed:28086084). Displays lower selectivity for K(+) over Na(+) ions (PubMed:28086084). Contributes to the native pacemaker currents in heart (If) and in the generation of the I(h) current which controls neuron excitability (PubMed:29936235, PubMed:30351409). Participates in cerebellar mechanisms of motor learning (By similarity). May mediate responses to sour stimuli (By similarity)
Cell membrane
Developmental and epileptic encephalopathy 24
A disease characterized by early-onset seizures, intellectual disability of varying degrees, and behavioral disturbances or autistic features in most individuals.
Catalyzes the hydrolysis of the 5-position phosphate of phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) and phosphatidylinositol-3,4,5-bisphosphate (PtdIns(3,4,5)P3), with the greatest catalytic activity towards PtdIns(4,5)P2 (PubMed:10764818, PubMed:15474001, PubMed:7761412, PubMed:9430698). Able also to hydrolyze the 5-phosphate of inositol 1,4,5-trisphosphate and of inositol 1,3,4,5-tetrakisphosphate (PubMed:25869668, PubMed:7761412). Regulates traffic in the endosomal pathway by regula
Cytoplasmic vesicle, phagosome membraneEarly endosome membraneMembrane, clathrin-coated pitCell projection, cilium, photoreceptor outer segmentCell projection, ciliumCytoplasmic vesicleEndosomeGolgi apparatus, trans-Golgi networkLysosome
Lowe oculocerebrorenal syndrome
X-linked multisystem disorder affecting eyes, nervous system, and kidney. It is characterized by hydrophthalmia, cataract, intellectual disability, vitamin D-resistant rickets, aminoaciduria, and reduced ammonia production by the kidney. Ocular abnormalities include cataract, glaucoma, microphthalmos, and decreased visual acuity. Developmental delay, hypotonia, behavior abnormalities, and areflexia are also present. Renal tubular involvement is characterized by impaired reabsorption of bicarbonate, amino acids, and phosphate. Musculoskeletal abnormalities such as joint hypermobility, dislocated hips, and fractures may develop as consequences of renal tubular acidosis and hypophosphatemia. Cataract is the only significant manifestation in carriers and is detected by slit-lamp examination.
Electrogenic sodium-dependent amino acid transporter with a preference for L-glutamine, L-alanine, L-histidine, L-aspartate and L-arginine. May facilitate glutamine uptake in both excitatory and inhibitory neurons. The transport mechanism and stoichiometry remain to be elucidated
MembraneCytoplasm, cell cortexCell projection, axon
Foveal hypoplasia 2
An isolated form of foveal hypoplasia, a developmental defect of the eye defined as the lack of foveal depression with continuity of all neurosensory retinal layers in the presumed foveal area. Clinical features include absence of foveal pit on optical coherence tomography, absence of foveal hyperpigmentation, absence of foveal avascularity, absence of foveal and macular reflexes, decreased visual acuity, and nystagmus. Optic nerve misrouting and anterior segment dysgenesis are observed in some FVH2 patients.
Voltage-gated potassium channel that mediates transmembrane potassium transport in excitable membranes, primarily in the brain and the central nervous system, but also in the cardiovascular system. Prevents aberrant action potential firing and regulates neuronal output. Forms tetrameric potassium-selective channels through which potassium ions pass in accordance with their electrochemical gradient. The channel alternates between opened and closed conformations in response to the voltage differen
Cell membraneMembraneCell projection, axonSynapseEndoplasmic reticulum membraneCell projection, lamellipodium membraneSynapse, synaptosomePresynaptic cell membraneCell projection, dendriteCell junction, paranodal septate junction
Developmental and epileptic encephalopathy 32
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE32 inheritance is autosomal dominant.
Associates with SLC3A1 to form a functional transporter complex that mediates the electrogenic exchange between cationic amino acids and neutral amino acids, with a stoichiometry of 1:1 (PubMed:16825196, PubMed:32494597, PubMed:32817565, PubMed:8663357). Has system b(0,+)-like activity with high affinity for extracellular cationic amino acids and L-cystine and lower affinity for intracellular neutral amino acids (PubMed:16825196, PubMed:32494597, PubMed:8663357). Substrate exchange is driven by
Apical cell membraneCell membrane
Cystinuria
An autosomal disorder characterized by impaired epithelial cell transport of cystine and dibasic amino acids (lysine, ornithine, and arginine) in the proximal renal tubule and gastrointestinal tract. The impaired renal reabsorption of cystine and its low solubility causes the formation of calculi in the urinary tract, resulting in obstructive uropathy, pyelonephritis, and, rarely, renal failure.
Cystine/H(+) symporter that mediates export of cystine, the oxidized dimer of cysteine, from lysosomes (PubMed:11689434, PubMed:15128704, PubMed:18337546, PubMed:22232659, PubMed:29467429, PubMed:33208952, PubMed:36113465). Plays an important role in melanin synthesis by catalyzing cystine export from melanosomes, possibly by inhibiting pheomelanin synthesis (PubMed:22649030). In addition to cystine export, also acts as a positive regulator of mTORC1 signaling in kidney proximal tubular cells, v
Lysosome membraneMelanosome membraneCell membrane
Cystinosis, nephropathic type
A form of cystinosis, a lysosomal storage disease due to defective transport of cystine across the lysosomal membrane. This results in cystine accumulation and crystallization in the cells causing widespread tissue damage. The classical nephropathic form has onset in the first year of life and is characterized by a polyuro-polydipsic syndrome, marked height-weight growth delay, generalized impaired proximal tubular reabsorptive capacity, with severe fluid-electrolyte balance alterations, renal failure, ocular symptoms and other systemic complications.
Beta subunit of the heteropentameric ligand-gated chloride channel gated by gamma-aminobutyric acid (GABA), a major inhibitory neurotransmitter in the brain (PubMed:10449790, PubMed:16412217, PubMed:26950270). GABA-gated chloride channels, also named GABA(A) receptors (GABAAR), consist of five subunits arranged around a central pore and contain one or two GABA active binding sites located at the alpha and beta subunit interfaces, depending on subunit composition (By similarity). When activated b
Postsynaptic cell membraneCell membrane
Developmental and epileptic encephalopathy 45
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent.
Translation elongation factor that catalyzes the GTP-dependent binding of aminoacyl-tRNA (aa-tRNA) to the A-site of ribosomes during the elongation phase of protein synthesis. Base pairing between the mRNA codon and the aa-tRNA anticodon promotes GTP hydrolysis, releasing the aa-tRNA from EEF1A1 and allowing its accommodation into the ribosome (By similarity). The growing protein chain is subsequently transferred from the P-site peptidyl tRNA to the A-site aa-tRNA, extending it by one amino acid
Endoplasmic reticulum membrane
Developmental and epileptic encephalopathy 33
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent.
Rod-specific cGMP phosphodiesterase that catalyzes the hydrolysis of 3',5'-cyclic GMP (PubMed:20940301). Necessary for the formation of a functional phosphodiesterase holoenzyme (By similarity). Involved in retinal circadian rhythm photoentrainment via modulation of UVA and orange light-induced phase-shift of the retina clock (By similarity). May participate in processes of transmission and amplification of the visual signal (PubMed:8394174)
MembraneCell projection, cilium, photoreceptor outer segment
Retinitis pigmentosa 40
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Catalytic subunit of the enzyme which catalyzes the decarboxylation of isocitrate (ICT) into alpha-ketoglutarate. The heterodimer composed of the alpha (IDH3A) and beta (IDH3B) subunits and the heterodimer composed of the alpha (IDH3A) and gamma (IDH3G) subunits, have considerable basal activity but the full activity of the heterotetramer (containing two subunits of IDH3A, one of IDH3B and one of IDH3G) requires the assembly and cooperative function of both heterodimers
Mitochondrion
Retinitis pigmentosa 90
A form of retinitis pigmentosa, a retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. RP90 is an autosomal recessive form.
Probable ATP-binding RNA helicase (Probable). Involved in pre-mRNA splicing as component of the spliceosome (PubMed:29301961, PubMed:9524131)
Nucleus
Retinitis pigmentosa 84
A form of retinitis pigmentosa, a retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. RP84 is an autosomal recessive, early onset form characterized by night blindness by age 4 and complete blindness by age 8. Funduscopy shows severely attenuated retinal vessels, severe macular atrophy, and prominent and deep macular colobomas lacking neuroretinal tissue.
Lysosomal acetyltransferase that acetylates the non-reducing terminal alpha-glucosamine residue of intralysosomal heparin or heparan sulfate, converting it into a substrate for luminal alpha-N-acetyl glucosaminidase
Lysosome membrane
Mucopolysaccharidosis 3C
A form of mucopolysaccharidosis type 3, an autosomal recessive lysosomal storage disease due to impaired degradation of heparan sulfate. MPS3 is characterized by severe central nervous system degeneration, but only mild somatic disease. Onset of clinical features usually occurs between 2 and 6 years; severe neurologic degeneration occurs in most patients between 6 and 10 years of age, and death occurs typically during the second or third decade of life.
Plays a role in pre-mRNA splicing as component of the U4/U6-U5 tri-snRNP complex that is involved in spliceosome assembly, and as component of the precatalytic spliceosome (spliceosome B complex)
NucleusNucleus speckle
Retinitis pigmentosa 70
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Potential calcium-dependent cell-adhesion protein. May be required for the structural integrity of the outer segment (OS) of photoreceptor cells (By similarity)
Cell membrane
Cone-rod dystrophy 15
An autosomal recessive retinal dystrophy characterized by retinal pigment deposits visible on fundus examination, predominantly in the macular region, and initial loss of cone photoreceptors followed by rod degeneration. This leads to decreased visual acuity and sensitivity in the central visual field, followed by loss of peripheral vision. Severe loss of vision occurs earlier than in retinitis pigmentosa, due to cone photoreceptors degenerating at a higher rate than rod photoreceptors.
May have a role in the excitatory ribbon synapse junctions between hair cells and cochlear ganglion cells and presumably also in analogous synapses within the retina
Cell membrane
Usher syndrome 3A
USH is a genetically heterogeneous condition characterized by the association of retinitis pigmentosa with sensorineural deafness. Age at onset and differences in auditory and vestibular function distinguish Usher syndrome type 1 (USH1), Usher syndrome type 2 (USH2) and Usher syndrome type 3 (USH3). USH3 is characterized by postlingual, progressive hearing loss, variable vestibular dysfunction, and onset of retinitis pigmentosa symptoms, including nyctalopia, constriction of the visual fields, and loss of central visual acuity, usually by the second decade of life.
With NUS1, forms the dehydrodolichyl diphosphate synthase (DDS) complex, an essential component of the dolichol monophosphate (Dol-P) biosynthetic machinery (PubMed:25066056, PubMed:28842490, PubMed:32817466, PubMed:33077723). Both subunits contribute to enzymatic activity, i.e. condensation of multiple copies of isopentenyl pyrophosphate (IPP) to farnesyl pyrophosphate (FPP) to produce dehydrodolichyl diphosphate (Dedol-PP), a precursor of dolichol phosphate which is utilized as a sugar carrier
Endoplasmic reticulum membrane
Retinitis pigmentosa 59
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Heterodimer with SLC3A2, that functions as an antiporter which operates as an efflux route by exporting cationic amino acids from inside the cells in exchange with neutral amino acids plus sodium ions and may participate in nitric oxide synthesis via the transport of L-arginine (PubMed:10080182, PubMed:10655553, PubMed:14603368, PubMed:15756301, PubMed:15776427, PubMed:17329401, PubMed:9829974, PubMed:9878049). Also mediates arginine transport in non-polarized cells, such as monocytes, and is es
Basolateral cell membrane
Lysinuric protein intolerance
A metabolic disorder characterized by increased renal excretion of cationic amino acid (CAA), reduced CAA absorption from intestine, and orotic aciduria. On a normal diet, LPI patients present poor feeding, vomiting, diarrhea, episodes of hyperammoniaemic coma and growth retardation. Hepatosplenomegaly, osteoporosis and a life-threatening pulmonary involvement (alveolar proteinosis) are also seen. Biochemically LPI is characterized by defective transport of dibasic amino acids at the basolateral membrane of epithelial cells in kidney and intestine.
Essential for primary ciliogenesis and embryonic development, facilitating the activation of Hedgehog (Hh) signaling pathway. Disrupts the interaction of GLI2 and GLI3 with the negative regulator SUFU. Inhibiting SUFU's interaction with GLI2 promotes the entry of GLI2 into the nucleus, allowing it to activate Hh target gene expression. Disrupting SUFU's interaction with GLI3 prevents its conversion into the repressor form, leading to increased nuclear GLI3 and enhanced Hh signaling. Required for
MembraneCytoplasm, cytoskeleton, cilium basal body
Joubert syndrome 2
A disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy and renal disease.
Metallocarboxypeptidase that mediates deglutamylation of tubulin and non-tubulin target proteins. Catalyzes the removal of polyglutamate side chains present on the gamma-carboxyl group of glutamate residues within the C-terminal tail of alpha- and beta-tubulin. Cleaves alpha- and gamma-linked polyglutamate tubulin side-chain, as well as the branching point glutamate. Also catalyzes the removal of alpha-linked glutamate residues from the carboxy-terminus of alpha-tubulin. Mediates deglutamylation
Cytoplasm, cytosolNucleusCytoplasm, cytoskeleton, spindleMidbody
Retinitis pigmentosa 75
A form of retinitis pigmentosa, a retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. RP75 inheritance is autosomal recessive.
Rod-specific cGMP phosphodiesterase that catalyzes the hydrolysis of 3',5'-cyclic GMP (PubMed:20940301). This protein participates in processes of transmission and amplification of the visual signal
Cell membraneCell projection, cilium, photoreceptor outer segment
Retinitis pigmentosa 43
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Critical isomerohydrolase in the retinoid cycle involved in regeneration of 11-cis-retinal, the chromophore of rod and cone opsins. Catalyzes the cleavage and isomerization of all-trans-retinyl fatty acid esters to 11-cis-retinol which is further oxidized by 11-cis retinol dehydrogenase to 11-cis-retinal for use as visual chromophore (PubMed:16116091). Essential for the production of 11-cis retinal for both rod and cone photoreceptors (PubMed:17848510). Also capable of catalyzing the isomerizati
CytoplasmCell membraneMicrosome membrane
Leber congenital amaurosis 2
A severe dystrophy of the retina, typically becoming evident in the first years of life. Visual function is usually poor and often accompanied by nystagmus, sluggish or near-absent pupillary responses, photophobia, high hyperopia and keratoconus.
Catalyzes the phosphorylation of hexose to hexose 6-phosphate, although at very low level compared to other hexokinases (PubMed:30517626). Has low glucose phosphorylating activity compared to other hexokinases (PubMed:30517626). Involved in glucose homeostasis and hepatic lipid accumulation. Required to maintain whole-body glucose homeostasis during pregnancy; however additional evidences are required to confirm this role (By similarity)
CytoplasmMitochondrion membranePhotoreceptor inner segment
Retinitis pigmentosa 92
A form of retinitis pigmentosa, a retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. RP92 is an autosomal recessive, mild form with onset of night blindness and vision loss in the third to sixth decades of life.
Together with ARL2, plays a role in the nuclear translocation, retention and transcriptional activity of STAT3. May play a role as an effector of ARL2
CytoplasmMitochondrion intermembrane spaceCytoplasm, cytoskeleton, microtubule organizing center, centrosomeNucleusCytoplasm, cytoskeleton, spindleCytoplasm, cytoskeleton, cilium basal body
Retinitis pigmentosa 82 with or without situs inversus
An autosomal recessive disorder characterized by variable association of retinitis pigmentosa with situs inversus. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. Situs inversus is a congenital abnormality in which organs in the thorax and the abdomen are opposite to their normal positions due to lateral transposition.
Plays a role in rod outer segment (ROS) morphogenesis (By similarity). May play a role with PRPH2 in the maintenance of the structure of ROS curved disks (By similarity). Plays a role in the organization of the ROS and maintenance of ROS disk diameter (By similarity). Involved in the maintenance of the retina outer nuclear layer (By similarity)
Photoreceptor inner segment membranePhotoreceptor outer segment membrane
Retinitis pigmentosa 7
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
May be involved in photoreceptor outer segment disk morphogenesis (By similarity)
CytoplasmPhotoreceptor inner segment
Cone-rod dystrophy 16
An inherited retinal dystrophy characterized by retinal pigment deposits visible on fundus examination, predominantly in the macular region, and initial loss of cone photoreceptors followed by rod degeneration. This leads to decreased visual acuity and sensitivity in the central visual field, followed by loss of peripheral vision. Severe loss of vision occurs earlier than in retinitis pigmentosa, due to cone photoreceptors degenerating at a higher rate than rod photoreceptors.
Chondroitin sulfate- and hyaluronan-binding proteoglycan involved in the organization of interphotoreceptor matrix; may participate in the maturation and maintenance of the light-sensitive photoreceptor outer segment. Binds heparin
Photoreceptor outer segment membranePhotoreceptor inner segment membraneSecreted, extracellular space, extracellular matrix, interphotoreceptor matrix
Retinitis pigmentosa 56
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Flippase that catalyzes in an ATP-dependent manner the transport of retinal-phosphatidylethanolamine conjugates like 11-cis and all-trans isomers of N-retinylidene-phosphatidylethanolamine (N-Ret-PE) from the lumen to the cytoplasmic leaflet of photoreceptor outer segment disk membranes, where 11-cis-retinylidene-phosphatidylethanolamine is then isomerized to its all-trans isomer and reduced by RDH8 to produce all-trans-retinol. This transport activity ensures that all-trans-retinal generated fr
MembraneEndoplasmic reticulumCytoplasmic vesicleCell projection, cilium, photoreceptor outer segment
Stargardt disease 1
An autosomal recessive form of Stargardt disease, a retinal degenerative disease characterized by macular dystrophy, progressive bilateral atrophy of the foveal retinal pigment epithelium, and accumulation of fluorescent flecks around the macula and/or in the central and near-peripheral areas of the retina. STGD1 patients typically lose central vision in their first or second decade of life.
Member of the ionotropic glutamate receptor family, which plays a crucial role in synaptic organization and signal transduction in the central nervous system. Although it shares structural features with ionotropic glutamate receptors, does not bind glutamate as a primary ligand (PubMed:34936451). Promotes synaptogenesis and mediates the D-Serine-dependent long term depression signals and AMPA receptor endocytosis of cerebellar parallel fiber-Purkinje cell (PF-PC) synapses through the NRX1B-CBLN1
Postsynaptic cell membrane
Spinocerebellar ataxia, autosomal recessive, 18
A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCAR18 features include progressive cerebellar atrophy, delayed psychomotor development, severely impaired gait, ocular movement abnormalities, and intellectual disability.
May be involved in modulating the expression of photoreceptor specific genes. Binds to the Ret-1 and Bat-1 element within the rhodopsin promoter
Nucleus
Macular degeneration, age-related, 6
A form of age-related macular degeneration, a multifactorial eye disease and the most common cause of irreversible vision loss in the developed world. In most patients, the disease is manifest as ophthalmoscopically visible yellowish accumulations of protein and lipid that lie beneath the retinal pigment epithelium and within an elastin-containing structure known as Bruch membrane.
Component of the IFT complex A (IFT-A), a complex required for retrograde ciliary transport and entry into cilia of G protein-coupled receptors (GPCRs) (PubMed:20889716, PubMed:22503633). Plays a pivotal role in proper development and function of ciliated cells through its role in ciliogenesis and/or cilium maintenance (PubMed:22503633). Required for the development and maintenance of the outer segments of rod and cone photoreceptor cells. Plays a role in maintenance and the delivery of opsin to
Cytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCell projection, cilium
Short-rib thoracic dysplasia 9 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome. SRTD9 is characterized by phalangeal cone-shaped epiphyses, chronic renal disease, nearly constant retinal dystrophy, and mild radiographic abnormality of the proximal femur. Occasional features include short stature, cerebellar ataxia, and hepatic fibrosis.
Plays an essential role for normal photoreceptor cell maintenance and vision
Cell projection, cilium, photoreceptor outer segmentPhotoreceptor inner segment
Retinitis pigmentosa 54
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Involved in pre-mRNA splicing as component of the spliceosome (PubMed:11867543, PubMed:20118938, PubMed:28781166). Required for the assembly of the U4/U5/U6 tri-snRNP complex, one of the building blocks of the spliceosome (PubMed:11867543)
NucleusNucleus speckleNucleus, Cajal body
Retinitis pigmentosa 11
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Acts as a guanine nucleotide exchange factor (GEF) for RhoA GTPases. Its activation induces formation of actin stress fibers. Also acts as a GEF for RAC1, inducing production of reactive oxygen species (ROS). Does not act as a GEF for CDC42. The G protein beta-gamma (Gbetagamma) subunits of heterotrimeric G proteins act as activators, explaining the integrated effects of LPA and other G-protein coupled receptor agonists on actin stress fiber formation, cell shape change and ROS production. Requi
CytoplasmCytoplasm, cytoskeletonCell membraneApical cell membrane
Retinitis pigmentosa 78
A form of retinitis pigmentosa, a retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. RP78 inheritance is autosomal recessive.
Centrosomal protein required for establishing a robust mitotic centrosome architecture that can endure the forces that converge on the centrosomes during spindle formation. Required for stabilizing the expanded pericentriolar material around the centriole
Cytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, cilium basal body
Retinitis pigmentosa 69
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Imports 4-aminobutanoate (GABA) into lysosomes. May act as a GABA sensor that regulates mTORC2-dependent INS signaling and gluconeogenesis. The transport mechanism and substrate selectivity remain to be elucidated
Lysosome membrane
Retinitis pigmentosa 68
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Involved in membrane protein trafficking at the base of the ciliary organelle. Mediates recruitment onto plasma membrane of the BBSome complex which would constitute a coat complex required for sorting of specific membrane proteins to the primary cilia (PubMed:20603001). Together with BBS1, is necessary for correct trafficking of PKD1 to primary cilia (By similarity). Together with the BBSome complex and LTZL1, controls SMO ciliary trafficking and contributes to the sonic hedgehog (SHH) pathway
Cell projection, cilium membraneCytoplasm, cytoskeleton, cilium axonemeCytoplasm, cytoskeleton, cilium basal body
Bardet-Biedl syndrome 3
A syndrome characterized by usually severe pigmentary retinopathy, early-onset obesity, polydactyly, hypogenitalism, renal malformation and intellectual disability. Secondary features include diabetes mellitus, hypertension and congenital heart disease. Bardet-Biedl syndrome inheritance is autosomal recessive, but three mutated alleles (two at one locus, and a third at a second locus) may be required for clinical manifestation of some forms of the disease.
Photoreceptor required for image-forming vision at low light intensity (PubMed:7846071, PubMed:8107847). Required for photoreceptor cell viability after birth (PubMed:12566452, PubMed:2215617). Light-induced isomerization of the chromophore 11-cis-retinal to all-trans-retinal triggers a conformational change that activates signaling via G-proteins (PubMed:26200343, PubMed:28524165, PubMed:28753425, PubMed:8107847). Subsequent receptor phosphorylation mediates displacement of the bound G-protein
MembraneCell projection, cilium, photoreceptor outer segment
Retinitis pigmentosa 4
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Involved in hearing and vision as member of the USH2 complex. In the inner ear, required for the maintenance of the hair bundle ankle formation, which connects growing stereocilia in developing cochlear hair cells. In retina photoreceptors, the USH2 complex is required for the maintenance of periciliary membrane complex that seems to play a role in regulating intracellular protein transport
Cell projection, stereocilium membraneSecreted
Usher syndrome 2A
USH is a genetically heterogeneous condition characterized by the association of retinitis pigmentosa with sensorineural deafness. Age at onset and differences in auditory and vestibular function distinguish Usher syndrome type 1 (USH1), Usher syndrome type 2 (USH2) and Usher syndrome type 3 (USH3). USH2 is characterized by congenital mild hearing impairment with normal vestibular responses.
Functions as an E3 ubiquitin-protein ligase and as an E3 SUMO1-protein ligase. Probable tumor suppressor involved in cell growth, cell proliferation and apoptosis that regulates p53/TP53 stability through ubiquitin-dependent degradation. May regulate chromatin modification through sumoylation of several chromatin modification-associated proteins. May be involved in DNA damage-induced cell death through IKBKE sumoylation
NucleusNucleus, PML body
Retinitis pigmentosa 31
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Variantes genéticas (ClinVar)
785 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
125 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Doença do transporte de aminoácidos
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Comparative cytotoxicity of novel mercury species α-mercuri-acetaldehyde and α-mercuri-acetic acid versus methylmercury in SH-SY5Y cells.
Recently, α-mercuri-acetaldehyde (HgCH2CHO) and α-mercuri-acetic acid (HgCH2COOH) have been proposed as potential causative agents of Minamata disease. However, their toxicological profiles remain largely unknown. This study aimed to characterize the cytotoxicity, cellular uptake, and efflux mechanisms of these compounds in SH-SY5Y neuroblastoma cells and to compare these properties with those of methylmercury (MeHg). Cell viability was assessed after 24 hr of exposure to MeHg (1-10 µM), HgCH2CHO (10-50 µM), or HgCH2COOH (10-50 µM) using the CCK-8 assay. The roles of L-type amino acid transporter 1 (LAT1) and multidrug resistance-associated proteins (MRPs) were evaluated using the inhibitors JPH203 (1 µM) and MK571 (10 µM), respectively. Intracellular mercury accumulation was quantified after 24 hr of exposure to 3 µM of each compound using thermal decomposition-amalgamation atomic absorption spectrometry. All compounds exhibited dose-dependent cytotoxicity, with a relative toxicity order of MeHg (LC50: 6.4 µM) > HgCH2CHO (LC50: 14.6 µM) > HgCH2COOH (LC50: 39.2 µM). LAT1 inhibition had minimal effect on MeHg toxicity but slightly attenuated that of HgCH2CHO and HgCH2COOH. Conversely, MRP inhibition markedly enhanced MeHg toxicity, modestly increased that of HgCH2CHO, and slightly increased that of HgCH2COOH. Cellular mercury accumulation was consistent with cytotoxicity patterns, showing 10-20-fold lower levels for HgCH2CHO and HgCH2COOH than for MeHg. HgCH2CHO and HgCH2COOH were approximately 2-5-fold less cytotoxic than MeHg and exhibited substantially lower intracellular mercury levels. Our findings suggest that HgCH2CHO and HgCH2COOH are unlikely to have neurotoxic potential comparable to that of MeHg.
TMAO converts cytochrome c into a pro-apoptotic peroxidase by destabilizing the heme-Met80 ligation.
Trimethylamine N-oxide (TMAO), a gut microbiota-derived metabolite, has been linked to cardiovascular, renal, and hepatic disorders, but its direct impact on mitochondrial apoptotic machinery remains unclear. Here, we show that TMAO binds cytochrome c (Cyt c), disrupting its structural integrity and converting it into an apoptotically competent species. Spectroscopic analyses revealed that TMAO destabilizes the heme-Met80 axial ligation, shifting Cyt c from its native hexacoordinate to a pentacoordinate state. This conformational change enhances peroxidase activity, exposes hydrophobic clusters, and perturbs the Trp microenvironment, marking Cyt c's transition from electron carrier to pro-apoptotic catalyst. Absorption spectra further showed splitting of the native 530 nm band into peaks at 520 and 550 nm, consistent with heme reduction. These alterations facilitate Cyt c release from the mitochondrial membrane and engagement in intrinsic apoptosis. Given that TMAO accumulates at higher concentrations in tissues enriched with oxygen transporters, such as kidney and liver, our findings provide mechanistic insight into its role in organ-specific toxicity, including chronic kidney disease (CKD) and non-alcoholic fatty liver disease (NAFLD). This study establishes a direct molecular link between TMAO and mitochondrial apoptosis via Cyt c destabilization, suggesting that stabilizing Cyt c could represent a therapeutic strategy against TMAO-associated pathologies.
Hartnup disease-causing SLC6A19 mutations lead to B0AT1 aberrant trafficking and ACE2 mis-localisation implicating the endoplasmic reticulum protein quality control.
The interaction between angiotensin-converting enzyme 2 (ACE2) and the sodium-dependent Broad neutral Amino acid Transporter 1 (B0AT1), encoded by the SLC6A19 gene, is increasingly recognized as pivotal in both physiological and pathological contexts. B0AT1 facilitates neutral amino acid transport and nutrient absorption, while ACE2 regulates vascular homeostasis and inflammation through the renin-angiotensin system. Mutations in SLC6A19 are implicated in Hartnup disease, a metabolic disorder characterized by defective amino acid transport. However, the cellular mechanisms underlying Hartnup disease-causing mutations' impact on B0AT1 and ACE2 function remain unclear. This study evaluated the subcellular localization and trafficking of 18 Hartnup disease-causing B0AT1 variants using experimental approaches including biochemical assays and In Silico analysis. The impact of these variants on ACE2 trafficking and plasma membrane targeting was also assessed to elucidate their interplay. Nine B0AT1 variants (R57C, G93R, R95P, R178Q, L242P, G284R, S303L, D517G, P579L) were found to be retained in the endoplasmic reticulum, impairing their trafficking to the plasma membrane. These variants were distributed across multiple B0AT1 structural domains. Importantly, several of these ER-retained variants, particularly R178Q and S303L, significantly disrupted ACE2 intracellular trafficking and its localization to the plasma membrane, indicating a direct effect on ACE2 subcellular targeting. The findings reveal that Hartnup disease-causing mutations can lead to ER retention of B0AT1, which in turn has a variable effect on ACE2 trafficking. This disruption likely contributes to Hartnup disease pathogenesis by impairing amino acid transport and may influence ACE2-mediated physiological functions beyond the renin-angiotensin system. Understanding these molecular mechanisms enhances insight into ACE2-B0AT1 interactions and could inform future therapeutic strategies and biomarker development for related disorders. Further research is needed to explore these pathways and their implications in disease.
Acute MK-801 induces hyperactivity and changes spatio-temporal exploratory dynamics without disrupting homebase retention in adult zebrafish.
The N-methyl-D-aspartate receptor (NMDAR) antagonist dizocilpine (MK-801) modulates locomotor functions and disrupts cognitive and spatial processes, making it useful for examining pharmacologically-induced behavioral phenotypes that mimic schizophrenia. Here, we investigated the impact of MK-801 on spatio-temporal exploratory dynamics in adult zebrafish, focusing on homebase behavior in the open field test (OFT). In Experiment 1, zebrafish received an intraperitoneal (i.p.) injection of saline or MK-801 (2.0 mg/kg), followed by a 15-min absorption period prior to a 30-min OFT. Experiment 2 involved an initial 30-min OFT, immediate saline or MK-801 (2.0 mg/kg), i.p. administration, and a retrial in the OFT 24 h later. Overall, MK-801 induced hyperactivity and stereotypy in Experiment 1. Although zebrafish were able to establish a homebase as a functionally relevant spatial reference, notable alterations in homebase-related behaviors were observed. Experiment 2 explored the homebase conservation across repeated OFT sessions, revealing that despite subtle changes in overall exploration and homebase occupancy, both groups demonstrated significant homebase conservation and reduced thigmotaxis in the retrial. An inhibitory avoidance task was also performed to confirm the amnesic effects of MK-801 on zebrafish (Experiment 3), revealing impaired aversive memory consolidation. Our novel findings indicate that while MK-801 altered movement patterns and disrupted aversive memory, zebrafish core spatial behaviors remained intact, highlighting the adaptive value of homebase as a conserved spatial strategy. Collectively, this work further supports the utility of zebrafish models for studying how pharmacological modulations of NMDAR affect spatial orientation and exploratory behavior, with translational relevance to neuropsychiatric diseases.
Pitfalls in the Diagnosis of Wilson Disease.
Wilson disease (WD), an uncommon autosomal recessive (AR) hereditary disorder characterized by abnormal accumulation of copper primarily in the liver and secondarily in other organs like the brain, is caused by a deficiency in the ATP7B transporter gene. The key to successful therapy is early diagnosis. Mutant genes need to be inherited from both parents for phenotypic expression. The ATP7B gene located on chromosome 13q14.3 comprises 20 introns and 21 exons, encodes a protein of 165 amino acids, and this helps in incorporation of copper into ceruloplasmin, the copper binding protein. So far, more than 800 mutations have been reported, of which 380 have confirmed involvement in the pathogenesis of WD. The most common mutations are H1069Q and R778L in European and Asian populations respectively. Approximately 90%-98% of WD subjects are heterozygous, showing different mutations in each of the alleles encoding the ATP7B. Conversely, the phenotype and the penetrance of WD can be extremely variable. Even patients carrying two disease-causing mutations do not necessarily have a demonstrable alteration of copper metabolism. Considering the possibility of late-onset disease, asymptomatic cases, and phenotypic variability, it is crucial to evaluate previous and future generations of the index case. WD ranges from being asymptomatic in some patients to result in acute liver failure and/or a variety of neuropsychiatric disorders among others. Although WD may be present at any age, is more common between the ages of 5 and 35 years. However, it should be investigated in patients with liver failure of unknown cause and those with liver disease associated with neuropsychiatric symptomatology. Diagnosis requires a combination of clinical signs and symptoms, as well as relevant diagnostic tests such as measurement of serum ceruloplasmin, urinary excretion of copper, liver biopsy or genetic testing. Treatment is lifelong and includes chelating agents (penicillamine and trientine) and inhibitors of copper absorption (zinc salts). Liver transplant is an option for patients with end-stage liver disease. The key to successful therapy is early diagnosis.
Publicações recentes
Intestinal transport of organic food compounds and drugs: A scoping review on the alterations observed in chronic kidney disease.
Sporisorium reilianum polysaccharides improve DSS-induced ulcerative colitis by regulating intestinal barrier function and metabolites.
Rebalancing of mitochondrial homeostasis through an NAD(+)-SIRT1 pathway preserves intestinal barrier function in severe malnutrition.
Anaplasma phagocytophilum Hijacks Flotillin and NPC1 Complex To Acquire Intracellular Cholesterol for Proliferation, Which Can Be Inhibited with Ezetimibe.
Novel Mechanistic PBPK Model to Predict Renal Clearance in Varying Stages of CKD by Incorporating Tubular Adaptation and Dynamic Passive Reabsorption.
📚 EuropePMCmostrando 199
Comparative cytotoxicity of novel mercury species α-mercuri-acetaldehyde and α-mercuri-acetic acid versus methylmercury in SH-SY5Y cells.
The Journal of toxicological sciencesA Rare Case of Congenital Glucose Galactose Malabsorption Due to SLC5A1 Mutation.
CureusTMAO converts cytochrome c into a pro-apoptotic peroxidase by destabilizing the heme-Met80 ligation.
Cellular and molecular biology (Noisy-le-Grand, France)Glucagon-like peptide-1 receptor agonists and risk for cardiovascular events in older adults treated with levothyroxine: a target trial emulation.
Cardiovascular diabetology[Exploring the potential causes of sarcopenia in sepsis patients based on proteome sequencing].
Zhonghua wei zhong bing ji jiu yi xueCase report: Lysine improvement in siblings with glutaric acidemia type 1 following reduced medical food intake: Implications for amino acid absorption and reabsorption.
Molecular genetics and metabolism reportsHartnup disease-causing SLC6A19 mutations lead to B0AT1 aberrant trafficking and ACE2 mis-localisation implicating the endoplasmic reticulum protein quality control.
Frontiers in cell and developmental biologyAcute MK-801 induces hyperactivity and changes spatio-temporal exploratory dynamics without disrupting homebase retention in adult zebrafish.
Behavioural brain researchAmino acids critical for lipid/s-interaction at the lipid-water-interface of TRPV5/TRPV6 remain different during vertebrate radiation: Relevance in cancer, bone disorders and other pathophysiologies.
Biochimica et biophysica acta. BiomembranesErgothioneine as a functional nutraceutical: Mechanisms, bioavailability, and therapeutic implications.
The Journal of nutritional biochemistryPitfalls in the Diagnosis of Wilson Disease.
Current neurology and neuroscience reportsSingle Amino Acid Supplementation in Inherited Metabolic Disorders: An Evidence-Based Review of Interventions.
GenesGly-βMCA modulates bile acid metabolism to reduce hepatobiliary injury in Mdr2 KO mice.
American journal of physiology. Gastrointestinal and liver physiologyBacteroides uniformis-generated hexadecanedioic acid ameliorates metabolic-associated fatty liver disease.
Gut microbesIdentification of a Novel NPC1L1 Inhibitor from Danshen and Its Role in Nonalcoholic Fatty Liver Disease.
International journal of molecular sciencesGestational exposure to polystyrene microplastics incurred placental damage in mice: Insights into metabolic and gene expression disorders.
Ecotoxicology and environmental safetyInherited disorders of vitamin metabolism.
European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology SocietyAromatic Amino Acid Metabolites: Molecular Messengers Bridging Immune-Microbiota Communication.
Immune networkTranscriptome and metabolome analysis reveal the mechanisms of iron absorption differences in apple rootstocks under alkaline condition.
Physiologia plantarumIntegrated Microbiome and Metabolome Analysis Reveals Correlations Between Gut Microbiota Components and Metabolic Profiles in Mice With Mitoxantrone-Induced Cardiotoxicity.
Drug design, development and therapyDesign of (R)-3-(5-Thienyl)carboxamido-2-aminopropanoic Acid Derivatives as Novel NMDA Receptor Glycine Site Agonists: Variation in Molecular Geometry to Improve Potency and Augment GluN2 Subunit-Specific Activity.
Journal of medicinal chemistryConversion of α-linolenic acid into n-3 long-chain polyunsaturated fatty acids: bioavailability and dietary regulation.
Critical reviews in food science and nutritionBanxia-Yiyiren alleviates insomnia and anxiety by regulating the gut microbiota and metabolites of PCPA-induced insomnia model rats.
Frontiers in microbiologySLC6A19 inhibition facilitates urinary neutral amino acid excretion and lowers plasma phenylalanine.
JCI insightIntestinal transport of organic food compounds and drugs: A scoping review on the alterations observed in chronic kidney disease.
Clinical nutrition ESPENMammalian colonic contribution of amino acids to whole-body homeostasis.
Current opinion in clinical nutrition and metabolic careExopeptidase combination enhances the degradation of isotopically labelled gluten immunogenic peptides in humans.
Frontiers in immunologyBacillus CotA laccase improved the intestinal health, amino acid metabolism and hepatic metabolic capacity of Pekin ducks fed naturally contaminated AFB1 diet.
Journal of animal science and biotechnologyDietary protein intake and the tubular handling of indoxyl sulfate.
Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal AssociationCauses of difficulties with adequate levothyroxine substitution - an immunoendocrine perspective.
Endokrynologia PolskaZearalenone-induced hepatointestinal toxicity in laying hens: unveiling the role of gut microbiota and fecal metabolites.
Poultry scienceMolecular Variability in Levodopa Absorption and Clinical Implications for the Management of Parkinson's Disease.
Journal of Parkinson's diseaseMolecular basis of inhibition of the amino acid transporter B0AT1 (SLC6A19).
Nature communicationsDietary Exposure to Acrylamide Has Negative Effects on the Gastrointestinal Tract: A Review.
NutrientsMultiomics reveals the ameliorating effect and underlying mechanism of aqueous extracts of polygonatum sibiricum rhizome on obesity and liver fat accumulation in high-fat diet-fed mice.
Phytomedicine : international journal of phytotherapy and phytopharmacologyInvestigation on the mechanisms of scorpion venom in hepatocellular carcinoma model mice via untargeted metabolomics profiling.
International immunopharmacologyImprovement of in vivo iron bioavailability using mung bean peptide-ferrous chelate.
Food research international (Ottawa, Ont.)Increased Absorption of Thyroxine in a Murine Model of Hypothyroidism Using Water/CO2 Nanobubbles.
International journal of molecular sciencesThe mitochondrial TSPO ligand Atriol mitigates metabolic-associated steatohepatitis by downregulating CXCL1.
Metabolism: clinical and experimentalComparative genomic and metabolomic analysis reveals the potential of a newly isolated Enterococcus faecium B6 involved in lipogenic effects.
GeneMulti-target Phenylpropanoids Against Epilepsy.
Current neuropharmacologyInflammation and Organic Cation Transporters Novel (OCTNs).
BiomoleculesN-acetylcysteine absorption and its potential dual effect improve fitness and fruit yield in Xylella fastidiosa infected plants.
Pest management scienceMyricanol improves metabolic profiles in dexamethasone induced lipid and protein metabolism disorders in mice.
Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapieMechanisms of Dangua Fang in multi-target and multi-method regulation of glycolipid metabolism based on phosphoproteomics.
Journal of traditional Chinese medicine = Chung i tsa chih ying wen panSporisorium reilianum polysaccharides improve DSS-induced ulcerative colitis by regulating intestinal barrier function and metabolites.
International journal of biological macromoleculesA comprehensive update on the ADMET considerations for α2δ calcium channel ligand medications for treating restless legs syndrome.
Expert opinion on drug metabolism & toxicologyRole of zinc in health and disease.
Clinical and experimental medicineImportant nutrient sources and carbohydrate metabolism patterns in the growth and development of spargana.
Parasites & vectorsVitamin K (Menadione)-incorporated chitosan/alginate hydrogel as a novel product for periorbital hyperpigmentation.
Journal of biomaterials science. Polymer editionTraditional Chinese Medicine formula Dai-Zong-Fang alleviating hepatic steatosis in db/db mice via gut microbiota modulation.
Frontiers in pharmacologyGut microbiota and metabolic profiles in chronic intermittent hypoxia-induced rats: disease-associated dysbiosis and metabolic disturbances.
Frontiers in endocrinologyA novel scenario in the therapeutic management of anemia of chronic kidney disease: placement and use of roxadustat.
Journal of nephrologyErgothioneine: an underrecognised dietary micronutrient required for healthy ageing?
The British journal of nutritionThe SLC6A15-SLC6A20 Neutral Amino Acid Transporter Subfamily: Functions, Diseases, and Their Therapeutic Relevance.
Pharmacological reviewsEvaluation of thiadiazine-based PET radioligands for imaging the AMPA receptor.
Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapieIntegration of 16S rRNA gene sequencing and LC/MS-based metabolomic analysis of early biomarkers of acute ischaemic stroke in Tibetan miniature pigs.
Journal of microbiological methodsIntegrated analysis of metabolomic and transcriptomic profiling reveals the effect of Atractylodes oil on Spleen Yang Deficiency Syndrome in rats.
Journal of ethnopharmacologyRebalancing of mitochondrial homeostasis through an NAD+-SIRT1 pathway preserves intestinal barrier function in severe malnutrition.
EBioMedicineSerum and urine metabolomics study revealed the amelioration of Gynura bicolor extract on high fat diet-fed and streptozotocin-induced type 2 diabetic mice based on UHPLC-MS/MS.
Journal of pharmaceutical and biomedical analysisRefractory Hypothyroidism: Unraveling the Complexities of Diagnosis and Management.
Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical EndocrinologistsAmino acids contribute to adaptive thermogenesis. New insights into the mechanisms of action of recent drugs for metabolic disorders are emerging.
Pharmacological researchLysinuric protein intolerance presenting as pancytopenia and splenomegaly mimicking acute leukaemia: a case report.
BMC pediatricsExploration of selected monoterpenes as potential TRPC channel family modulator in lung cancer, an in-silico upshot.
Journal of biomolecular structure & dynamicsAn in-silico investigation based on molecular simulations of novel and potential brain-penetrant GluN2B NMDA receptor antagonists as anti-stroke therapeutic agents.
Journal of biomolecular structure & dynamicsA novel SLC5A6 homozygous variant in a family with multivitamin-dependent neurometabolic disorder: Phenotype expansion and long-term follow-up.
European journal of medical geneticsA combined proteomics and metabolomics analysis reveals the invisible regulation of plant root responses to oxybenzone (benzophenone-3) stress.
The Science of the total environmentComprehensive expression analysis reveals several miRNAs against acute pancreatitis via modulating autophagy.
Cellular and molecular biology (Noisy-le-Grand, France)Comprehensive Proteome and Acetyl-Proteome Atlas Reveals Hepatic Lipid Metabolism in Layer Hens with Fatty Liver Hemorrhagic Syndrome.
International journal of molecular sciencesMolecular mechanisms involved in fetal programming and disease origin in adulthood.
Journal of pediatric endocrinology & metabolism : JPEMApolipoprotein E (APOE) Haplotypes in Healthy Subjects from Worldwide Macroareas: A Population Genetics Perspective for Cardiovascular Disease, Neurodegeneration, and Dementia.
Current issues in molecular biologyMilk protects against sarcopenic obesity due to increase in the genus Akkermansia in faeces of db/db mice.
Journal of cachexia, sarcopenia and muscleNew aspects for the brain in Hartnup disease based on mining of high-resolution cellular mRNA expression data for SLC6A19.
IBRO neuroscience reportsThe Influence of Alcohol Consumption on Intestinal Nutrient Absorption: A Comprehensive Review.
NutrientsTemporal Proteomic and Lipidomic Profiles of Cerulein-Induced Acute Pancreatitis Reveal Novel Insights for Metabolic Alterations in the Disease Pathogenesis.
ACS omegaCecal Microbial Succession and Its Apparent Association with Nutrient Metabolism in Broiler Chickens.
mSphereTherapeutic effect and mechanism of Daikenchuto in a model of methotrexate-induced acute small intestinal mucositis.
PloS oneDietary advanced glycation end products (dAGEs): An insight between modern diet and health.
Food chemistryGastrointestinal barriers to levodopa transport and absorption in Parkinson's disease.
European journal of neurologyVirtual screening and activity evaluation of human uric acid transporter 1 (hURAT1) inhibitors.
RSC advancesWilson's disease: overview.
Medicina clinicaContributions of SGK3 to transporter-related diseases.
Frontiers in cell and developmental biologyEnhancing Mechanisms of the Plant Growth-Promoting Bacterial Strain Brevibacillus sp. SR-9 on Cadmium Enrichment in Sweet Sorghum by Metagenomic and Transcriptomic Analysis.
International journal of environmental research and public healthCOVID-19 Vaccines and the Virus: Impact on Drug Metabolism and Pharmacokinetics.
Drug metabolism and disposition: the biological fate of chemicalsProteome Analysis of Temporomandibular Joint with Disc Displacement.
Journal of dental researchExcretion of excess nitrogen and increased survival by loss of SLC6A19 in a mouse model of ornithine transcarbamylase deficiency.
Journal of inherited metabolic diseaseSystemic tryptophan homeostasis.
Frontiers in molecular biosciencesNontargeted metabolomics to characterize the effects of isotretinoin on skin metabolism in rabbit with acne.
Frontiers in pharmacologyAristolochic acid-induced nephropathy is attenuated in mice lacking the neutral amino acid transporter B0AT1 (Slc6a19).
American journal of physiology. Renal physiologySynthesis of 2-Acetamido-1,3,4-Tri-O-Acetyl-2-Deoxy-D-Mannopyranose -6-Phosphate Prodrugs as Potential Therapeutic Agents.
Current protocolsAn emerging role of vitamin D3 in amino acid absorption in different intestinal segments of on-growing grass carp (Ctenopharyngodon idella).
Animal nutrition (Zhongguo xu mu shou yi xue hui)Nutritional deficiency in an intestine-on-a-chip recapitulates injury hallmarks associated with environmental enteric dysfunction.
Nature biomedical engineeringMetabolomics Study Suggests the Mechanism of Different Types of Tieguanyin (Oolong) Tea in Alleviating Alzheimer's Disease in APP/PS1 Transgenic Mice.
MetabolitesClinical and Genetic Analysis of a Family With Sitosterolemia Caused by a Novel ATP-Binding Cassette Subfamily G Member 5 Compound Heterozygous Mutation.
Frontiers in cardiovascular medicineDiosgenin Ameliorates Non-alcoholic Fatty Liver Disease by Modulating the Gut Microbiota and Related Lipid/Amino Acid Metabolism in High Fat Diet-Fed Rats.
Frontiers in pharmacologyFunctional roles of taurine, L-theanine, L-citrulline, and betaine during heat stress in poultry.
Journal of animal science and biotechnologyModulation of Naturally Occurring Linear Dipeptide Chirality to Reduce the Affinity for Oligopeptide Transporter 1 and Increase Intestinal Stability for an Enhanced Colon-Targeting Effect in the Treatment of Inflammatory Bowel Disease: An Application of trans-4-l-Hydroxyprolyl-l-serine.
Journal of medicinal chemistryChandamarutha Chenduram, an Indian traditional Siddha preparation attenuated the neuronal degeneration in ischemic mice through ameliorating cytokines and oxy-radicals mediated EAAT-2 dysfunction.
Journal of ethnopharmacologyFluorinated dihydropyridines as candidates to block L-type voltage-dependent calcium channels.
Journal of biomolecular structure & dynamicsRecent advances in riboflavin transporter RFVT and its genetic disease.
Pharmacology & therapeuticsAnaplasma phagocytophilum Hijacks Flotillin and NPC1 Complex To Acquire Intracellular Cholesterol for Proliferation, Which Can Be Inhibited with Ezetimibe.
mBioGenomic and epigenomic evolution of acquired resistance to combination therapy in esophageal squamous cell carcinoma.
JCI insightDecreased Expression of Ileal Thyroid Hormone Transporters in a Hypothyroid Patient: A Case Report.
Frontiers in endocrinologySimulation of Remdesivir Pharmacokinetics and Its Drug Interactions.
Journal of pharmacy & pharmaceutical sciences : a publication of the Canadian Society for Pharmaceutical Sciences, Societe canadienne des sciences pharmaceutiquesUnique Regulation of Coupled NaCl Absorption by Inducible Nitric Oxide in a Spontaneous SAMP1/YitFc Mouse Model of Chronic Intestinal Inflammation.
Inflammatory bowel diseasesCombined ASBT Inhibitor and FGF15 Treatment Improves Therapeutic Efficacy in Experimental Nonalcoholic Steatohepatitis.
Cellular and molecular gastroenterology and hepatologyEncephalitis with Autoantibodies against the Glutamate Kainate Receptors GluK2.
Annals of neurologyZinc transporter mutations linked to acrodermatitis enteropathica disrupt function and cause mistrafficking.
The Journal of biological chemistryAn Update on Evaluation and Management in Cystinuria.
UrologyATB0,+-targeted delivery of triptolide prodrugs for safer and more effective pancreatic cancer therapy.
Bioorganic & medicinal chemistry lettersClinical and molecular characterization of the R751L-CFTR mutation.
American journal of physiology. Lung cellular and molecular physiologyEncapsulated crystalline lysine and DL-methionine have higher efficiency than the crystalline form in broilers.
Poultry scienceACE2 and gut amino acid transport.
Clinical science (London, England : 1979)Hybridization of Curcumin Analogues with Cinnamic Acid Derivatives as Multi-Target Agents Against Alzheimer's Disease Targets.
Molecules (Basel, Switzerland)Unraveling the Role of ACE2, the Binding Receptor for SARS-CoV-2, in Inflammatory Bowel Disease.
Inflammatory bowel diseasesNovel Mechanistic PBPK Model to Predict Renal Clearance in Varying Stages of CKD by Incorporating Tubular Adaptation and Dynamic Passive Reabsorption.
CPT: pharmacometrics & systems pharmacologyUse of encapsulated L-lysine-HCl and DL-methionine improves postprandial amino acid balance in laying hens.
Journal of animal scienceEffects of titanium dioxide nanoparticles on nutrient absorption and metabolism in rats: distinguishing the susceptibility of amino acids, metal elements, and glucose.
NanotoxicologyMembrane Transporters for Amino Acids as Players of Cancer Metabolic Rewiring.
CellsThe leaves of Bougainvillea spectabilis suppressed inflammation and nociception in vivo through the modulation of glutamatergic, cGMP, and ATP-sensitive K+ channel pathways.
Journal of ethnopharmacologyPostnatal growth retardation is associated with intestinal mucosa mitochondrial dysfunction and aberrant energy status in piglets.
Journal of cellular and molecular medicineNonclinical Characterization of the Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitor Roxadustat, a Novel Treatment of Anemia of Chronic Kidney Disease.
The Journal of pharmacology and experimental therapeuticsCinnamaldehyde improves the growth performance and digestion and absorption capacity in grass carp (Ctenopharyngodon idella).
Fish physiology and biochemistryProteomic and metabolomic characterization of cardiac tissue in acute myocardial ischemia injury rats.
PloS oneHepcidin and Erythroferrone Correlate with Hepatic Iron Transporters in Rats Supplemented with Multispecies Probiotics.
Molecules (Basel, Switzerland)In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel.
Journal of biomolecular structure & dynamicsThe dynamic process of dietary soybean β-conglycinin in digestion, absorption, and metabolism among different intestinal segments in grass carp (Ctenopharyngodon idella).
Fish physiology and biochemistryIntestinal Permeability and Oral Absorption of Selected Drugs Are Reduced in a Mouse Model of Familial Alzheimer's Disease.
Molecular pharmaceuticsDiscovery of Potential Species-Specific Green Insecticides Targeting the Lepidopteran Ryanodine Receptor.
Journal of agricultural and food chemistryAn unusually high plasma concentration of homocysteine resulting from a combination of so-called "secondary" etiologies.
Clinical biochemistryMechanisms of Regulation of Transporters of Amino Acid Absorption in Inflammatory Bowel Diseases.
Comprehensive PhysiologyAttenuation of the Hepatoprotective Effects of Ileal Apical Sodium Dependent Bile Acid Transporter (ASBT) Inhibition in Choline-Deficient L-Amino Acid-Defined (CDAA) Diet-Fed Mice.
Frontiers in medicineEffects of glucocorticoids on the gene expression of nutrient transporters in different rabbit intestinal segments.
Animal : an international journal of animal bioscienceDietary protein-bound or free amino acids differently affect intestinal morphology, gene expression of amino acid transporters, and serum amino acids of pigs exposed to heat stress.
Journal of animal scienceAnalysis of the relationship between the mutation site of the SLC39A4 gene and acrodermatitis enteropathica by reporting a rare Chinese twin: a case report and review of the literature.
BMC pediatricsComputational Modeling Explains the Multi Sterol Ligand Specificity of the N-Terminal Domain of Niemann-Pick C1-Like 1 Protein.
ACS omegay+LAT1 and y+LAT2 contribution to arginine uptake in different human cell models: Implications in the pathophysiology of Lysinuric Protein Intolerance.
Journal of cellular and molecular medicineContributions of Gut Bacteria and Diet to Drug Pharmacokinetics in the Treatment of Parkinson's Disease.
Frontiers in neurologyEffect of Euphorbia factor L1 on intestinal barrier impairment and defecation dysfunction in Caenorhabditis elegans.
Phytomedicine : international journal of phytotherapy and phytopharmacologyImproving Eflornithine Oral Bioavailability and Brain Uptake by Modulating Intercellular Junctions With an E-cadherin Peptide.
Journal of pharmaceutical sciencesMice Lacking the Intestinal and Renal Neutral Amino Acid Transporter SLC6A19 Demonstrate the Relationship between Dietary Protein Intake and Amino Acid Malabsorption.
NutrientsStructure and mechanism of the cation-chloride cotransporter NKCC1.
NatureMetabonomics of mice intestine in Codonopsis foetens induced apoptosis of intestine cancer cells.
Saudi journal of biological sciencesL-Tryptophan represses persister formation via inhibiting bacterial motility and promoting antibiotics absorption.
Future microbiologySoybean glycinin decreased growth performance, impaired intestinal health, and amino acid absorption capacity of juvenile grass carp (Ctenopharyngodon idella).
Fish physiology and biochemistryTranscriptomic analysis of enteropathy in Zambian children with severe acute malnutrition.
EBioMedicineEffect of renal tubule-specific knockdown of the Na+/H+ exchanger NHE3 in Akita diabetic mice.
American journal of physiology. Renal physiologyN-Carbamylglutamate and l-Arginine Promote Intestinal Absorption of Amino Acids by Regulating the mTOR Signaling Pathway and Amino Acid and Peptide Transporters in Suckling Lambs with Intrauterine Growth Restriction.
The Journal of nutritionBioavailability and metabolism of selected cocoa bioactive compounds: A comprehensive review.
Critical reviews in food science and nutrition[Elucidation of Disease Mechanisms Based on Transport Function at Tissue Barriers and Challenges in Drug Development].
Yakugaku zasshi : Journal of the Pharmaceutical Society of JapanA new era for migraine: Pharmacokinetic and pharmacodynamic insights into monoclonal antibodies with a focus on galcanezumab, an anti-CGRP antibody.
Cephalalgia : an international journal of headacheBacillus amyloliquefaciens Ameliorates H2O2-Induced Oxidative Damage by Regulating Transporters, Tight Junctions, and Apoptosis Gene Expression in Cell Line IPEC-1.
Probiotics and antimicrobial proteinsSubstitutions that lock and unlock the proton-coupled folate transporter (PCFT-SLC46A1) in an inward-open conformation.
The Journal of biological chemistryEffects of chronic stress on intestinal amino acid pathways.
Physiology & behaviorInhibition of Niemann-Pick C1-Like 1 by Ezetimibe Reduces Dietary 5β,6β-Epoxycholesterol Absorption in Rats.
Cardiovascular drugs and therapyAmino Acid Transport Across the Mammalian Intestine.
Comprehensive PhysiologyGastrointestinal Malabsorption of Thyroxine.
Endocrine reviewsDevelopment of Biomarkers for Inhibition of SLC6A19 (B⁰AT1)-A Potential Target to Treat Metabolic Disorders.
International journal of molecular sciencesEvaluation of the Mitra microsampling device for use with key urinary metabolites in patients with Alkaptonuria.
BioanalysisBasolateral presence of the proinflammatory cytokine tumor necrosis factor -α and secretions from adipocytes and macrophages reduce intestinal sugar transport.
Journal of cellular physiologyCyclooxygenase pathway mediates the inhibition of Na-glutamine co-transporter B0AT1 in rabbit villus cells during chronic intestinal inflammation.
PloS oneResveratrol enhances cisplatin-induced apoptosis in human hepatoma cells via glutamine metabolism inhibition.
BMB reportsInhibiting neutral amino acid transport for the treatment of phenylketonuria.
JCI insightImproving the diagnosis of cobalamin and related defects by genomic analysis, plus functional and structural assessment of novel variants.
Orphanet journal of rare diseasesCholera toxin inhibits SNX27-retromer-mediated delivery of cargo proteins to the plasma membrane.
Journal of cell scienceAdenosinergic signaling inhibits oxalate transport by human intestinal Caco2-BBE cells through the A2B adenosine receptor.
American journal of physiology. Cell physiologyScaling behavior of drug transport and absorption in in silico cerebral capillary networks.
PloS oneRabbit SLC15A1, SLC7A1 and SLC1A1 genes are affected by site of digestion, stage of development and dietary protein content.
Animal : an international journal of animal bioscienceStructural insights into pharmacophore-assisted in silico identification of protein-protein interaction inhibitors for inhibition of human toll-like receptor 4 - myeloid differentiation factor-2 (hTLR4-MD-2) complex.
Journal of biomolecular structure & dynamicsProton Coupled Oligopeptide Transporter 1 (PepT1) Function, Regulation, and Influence on the Intestinal Homeostasis.
Comprehensive PhysiologyFunction, Regulation, and Pathophysiological Relevance of the POT Superfamily, Specifically PepT1 in Inflammatory Bowel Disease.
Comprehensive PhysiologyHigh-fat diet affects gut nutrients transporters in hypo and hyperthyroid mice by PPAR-a independent mechanism.
Life sciencesCooperation of Antiporter LAT2/CD98hc with Uniporter TAT1 for Renal Reabsorption of Neutral Amino Acids.
Journal of the American Society of Nephrology : JASNThe Pharmabiotic Approach to Treat Hyperammonemia.
NutrientsEffects of dietary supplementation of arginine-silicate-inositol complex on absorption and metabolism of calcium of laying hens.
PloS oneHereditary folate malabsorption due to a mutation in the external gate of the proton-coupled folate transporter SLC46A1.
Blood advancesDelivery of Oxytocin to the Brain for the Treatment of Autism Spectrum Disorder by Nasal Application.
Molecular pharmaceuticsDopamine and Levodopa Prodrugs for the Treatment of Parkinson's Disease.
Molecules (Basel, Switzerland)Varying the ratio of Lys:Met while maintaining the ratios of Thr:Phe, Lys:Thr, Lys:His, and Lys:Val alters mammary cellular metabolites, mammalian target of rapamycin signaling, and gene transcription.
Journal of dairy scienceInteraction of Excitatory Amino Acid Transporters 1 - 3 (EAAT1, EAAT2, EAAT3) with N-Carbamoylglutamate and N-Acetylglutamate.
Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacologyEffect of early weaning on the expression of excitatory amino acid transporter 1 in the jejunum and ileum of piglets.
Molecular medicine reportsA review of patient-specific gastrointestinal parameters as a platform for developing in vitro models for predicting the in vivo performance of oral dosage forms in patients with Parkinson's disease.
International journal of pharmaceuticsOxidative species-induced excitonic transport in tubulin aromatic networks: Potential implications for neurodegenerative disease.
Journal of photochemistry and photobiology. B, BiologyTherapeutic Effect of Astroglia-like Mesenchymal Stem Cells Expressing Glutamate Transporter in a Genetic Rat Model of Depression.
TheranosticsEnhancement of nose-to-brain delivery of hydrophilic macromolecules with stearate- or polyethylene glycol-modified arginine-rich peptide.
International journal of pharmaceuticsAmino Acid Hydration Decreases Radiation-Induced Nausea in Mice: A Pica Model.
Advances in experimental medicine and biologyAcute Neuroinflammation Promotes Cell Responses to 1800 MHz GSM Electromagnetic Fields in the Rat Cerebral Cortex.
Neurotoxicity research[Quantitative Targeted Absolute Proteomics (QTAP)-based Pharmacoproteomics: The Importance of International Collaboration].
Yakugaku zasshi : Journal of the Pharmaceutical Society of JapanMemory and Learning Dysfunction Following Copper Toxicity: Biochemical and Immunohistochemical Basis.
Molecular neurobiologyA common NHE3 single-nucleotide polymorphism has normal function and sensitivity to regulatory ligands.
American journal of physiology. Gastrointestinal and liver physiologyThe Effect of Uremic Solutes on the Organic Cation Transporter 2.
Journal of pharmaceutical sciencesAltered cargo proteins of human plasma endothelial cell-derived exosomes in atherosclerotic cerebrovascular disease.
FASEB journal : official publication of the Federation of American Societies for Experimental BiologyRiboflavin Responsive Mitochondrial Dysfunction in Neurodegenerative Diseases.
Journal of clinical medicine[Influence of occupational aluminum exposure on cognitive function and glutamate receptor protein expression in workers].
Zhonghua lao dong wei sheng zhi ye bing za zhi = Zhonghua laodong weisheng zhiyebing zazhi = Chinese journal of industrial hygiene and occupational diseasesHypoxia-inducible factor-1α activation improves renal oxygenation and mitochondrial function in early chronic kidney disease.
American journal of physiology. Renal physiologyPhenylalanine isotope pulse method to measure effect of sepsis on protein breakdown and membrane transport in the pig.
American journal of physiology. Endocrinology and metabolismMYC-driven inhibition of the glutamate-cysteine ligase promotes glutathione depletion in liver cancer.
EMBO reportsKinetics, subcellular localization, and contribution to parasite virulence of a Trypanosoma cruzi hybrid type A heme peroxidase (TcAPx-CcP).
Proceedings of the National Academy of Sciences of the United States of AmericaKidney Cadmium Toxicity, Diabetes and High Blood Pressure: The Perfect Storm.
The Tohoku journal of experimental medicineAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Doença do transporte de aminoácidos.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Doença do transporte de aminoácidos
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Comparative cytotoxicity of novel mercury species α-mercuri-acetaldehyde and α-mercuri-acetic acid versus methylmercury in SH-SY5Y cells.
- TMAO converts cytochrome c into a pro-apoptotic peroxidase by destabilizing the heme-Met80 ligation.
- Hartnup disease-causing SLC6A19 mutations lead to B0AT1 aberrant trafficking and ACE2 mis-localisation implicating the endoplasmic reticulum protein quality control.
- Acute MK-801 induces hyperactivity and changes spatio-temporal exploratory dynamics without disrupting homebase retention in adult zebrafish.
- Pitfalls in the Diagnosis of Wilson Disease.
- Intestinal transport of organic food compounds and drugs: A scoping review on the alterations observed in chronic kidney disease.
- Sporisorium reilianum polysaccharides improve DSS-induced ulcerative colitis by regulating intestinal barrier function and metabolites.
- Rebalancing of mitochondrial homeostasis through an NAD(+)-SIRT1 pathway preserves intestinal barrier function in severe malnutrition.
- Anaplasma phagocytophilum Hijacks Flotillin and NPC1 Complex To Acquire Intracellular Cholesterol for Proliferation, Which Can Be Inhibited with Ezetimibe.
- Novel Mechanistic PBPK Model to Predict Renal Clearance in Varying Stages of CKD by Incorporating Tubular Adaptation and Dynamic Passive Reabsorption.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:79166(Orphanet)
- MONDO:0019216(MONDO)
- GARD:18948(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q471778(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
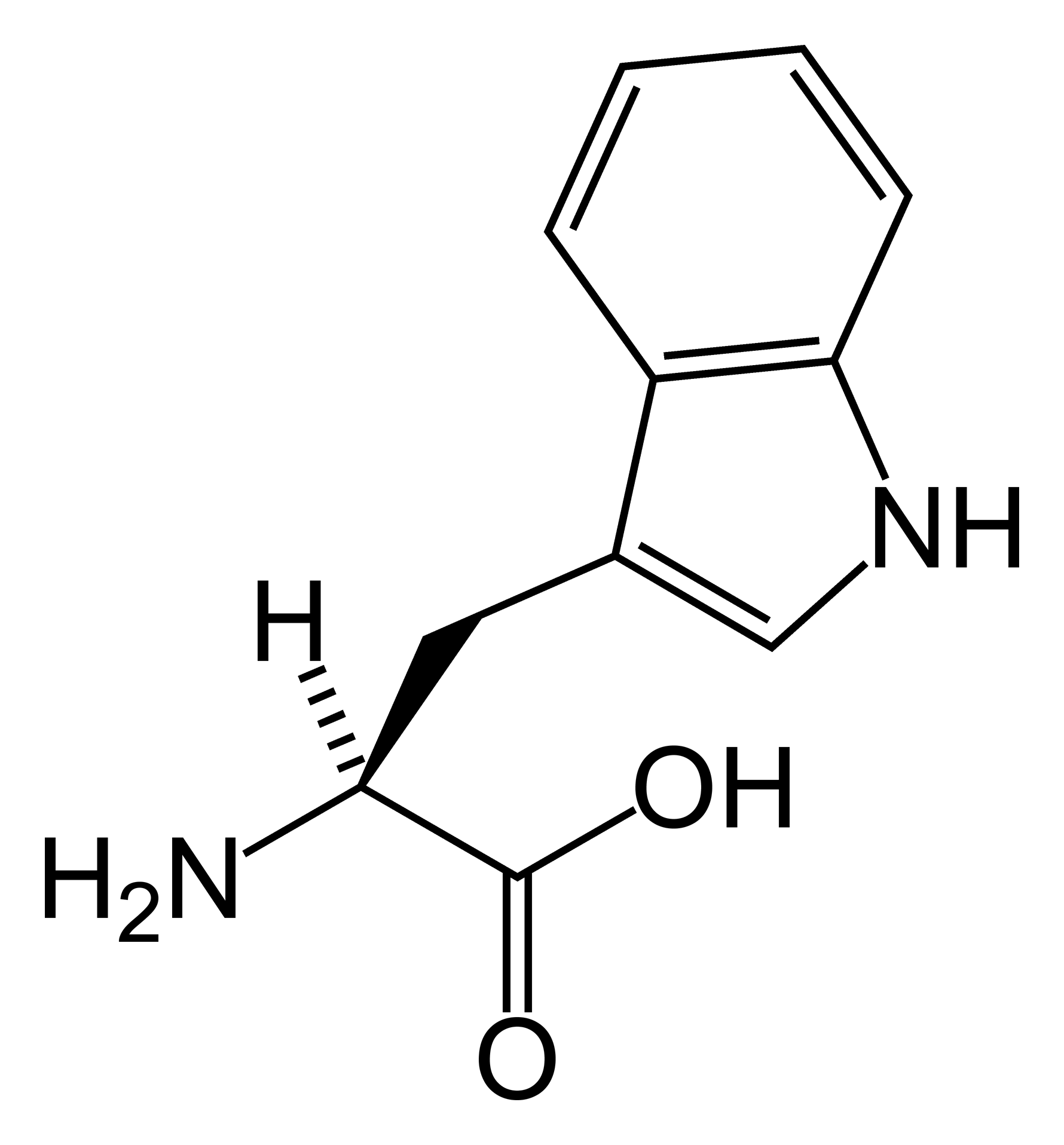
Doença do transporte de aminoácidos
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata