Distúrbio hereditário que afeta o metabolismo de qualquer composto ácido contendo carbono em uma ligação covalente.
Introdução
O que você precisa saber de cara
Distúrbio hereditário que afeta o metabolismo de qualquer composto ácido contendo carbono em uma ligação covalente.
Tem tratamento?
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 249 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 578 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Triagem neonatal (Teste do Pezinho)
A triagem neonatal permite diagnóstico precoce e início imediato do tratamento.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
43 genes identificados com associação a esta condição.
May play some role in mitochondrial processes
Mitochondrion
3-methylglutaconic aciduria 3
An autosomal recessive metabolic disorder that causes a neuro-ophthalmologic syndrome consisting of early-onset bilateral optic atrophy, spasticity, extrapyramidal dysfunction and cognitive deficit. Urinary excretion of 3-methylglutaconic acid and 3-methylglutaric acid is increased. MGCA3 can be distinguished from MGCA1 by the absence of increase of 3-hydroxyisovaleric acid levels.
Catalyzes the fifth step in the leucine degradation pathway, the reversible hydration of 3-methylglutaconyl-CoA (3-MG-CoA) to 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) (PubMed:11738050, PubMed:12434311, PubMed:12655555, PubMed:16640564). Can catalyze the reverse reaction but at a much lower rate in vitro (PubMed:16640564). HMG-CoA is then quickly degraded by another enzyme (such as HMG-CoA lyase) to give acetyl-CoA and acetoacetate (PubMed:16640564). Uses other substrates such as (2E)-glutaconyl-
Mitochondrion
3-methylglutaconic aciduria 1
An inborn error of leucine metabolism. It leads to an autosomal recessive syndrome with variable clinical phenotype, ranging from delayed speech development to severe psychomotor retardation, coma, failure to thrive, metabolic acidosis and dystonia. MGCA1 can be distinguished from other forms of MGCA by the pattern of metabolite excretion: 3-methylglutaconic acid levels are higher than those detected in other forms, whereas methylglutaric acid levels are usually only slightly elevated and there is a high level of 3-hydroxyisovaleric acid excretion (not present in other MGCA forms).
This is one of the 2 subunits of the biotin-dependent propionyl-CoA carboxylase (PCC), a mitochondrial enzyme involved in the catabolism of odd chain fatty acids, branched-chain amino acids isoleucine, threonine, methionine, and valine and other metabolites (PubMed:15890657, PubMed:6765947). Propionyl-CoA carboxylase catalyzes the carboxylation of propionyl-CoA/propanoyl-CoA to D-methylmalonyl-CoA/(S)-methylmalonyl-CoA (PubMed:15890657, PubMed:6765947). Within the holoenzyme, the alpha subunit c
Mitochondrion matrix
Propionic acidemia type II
Life-threatening disease characterized by episodic vomiting, lethargy and ketosis, neutropenia, periodic thrombocytopenia, hypogammaglobulinemia, developmental retardation, and intolerance to protein.
Mitochondrial 3-hydroxy-3-methylglutaryl-CoA lyase that catalyzes a cation-dependent cleavage of (S)-3-hydroxy-3-methylglutaryl-CoA into acetyl-CoA and acetoacetate, a key step in ketogenesis. Terminal step in leucine catabolism. Ketone bodies (beta-hydroxybutyrate, acetoacetate and acetone) are essential as an alternative source of energy to glucose, as lipid precursors and as regulators of metabolism
Mitochondrion matrixPeroxisome
3-hydroxy-3-methylglutaryl-CoA lyase deficiency
An autosomal recessive disease affecting ketogenesis and L-leucine catabolism. The disease usually appears in the first year of life after a fasting period and its clinical acute symptoms include vomiting, seizures, metabolic acidosis, hypoketotic hypoglycemia and lethargy. These symptoms sometimes progress to coma, with fatal outcome in some cases.
Membrane
The branched-chain alpha-keto dehydrogenase complex catalyzes the overall conversion of alpha-keto acids to acyl-CoA and CO(2). It contains multiple copies of three enzymatic components: branched-chain alpha-keto acid decarboxylase (E1), lipoamide acyltransferase (E2) and lipoamide dehydrogenase (E3). Within this complex, the catalytic function of this enzyme is to accept, and to transfer to coenzyme A, acyl groups that are generated by the branched-chain alpha-keto acid decarboxylase component
Mitochondrion matrix
Biotin-attachment subunit of the 3-methylcrotonyl-CoA carboxylase, an enzyme that catalyzes the conversion of 3-methylcrotonyl-CoA to 3-methylglutaconyl-CoA, a critical step for leucine and isovaleric acid catabolism
Mitochondrion matrix
3-methylcrotonoyl-CoA carboxylase 1 deficiency
An autosomal recessive disorder of leucine catabolism. The phenotype is variable, ranging from neonatal onset with severe neurological involvement to asymptomatic adults. There is a characteristic organic aciduria with massive excretion of 3-hydroxyisovaleric acid and 3-methylcrotonylglycine, usually in combination with a severe secondary carnitine deficiency.
Thiol-specific peroxidase that catalyzes the reduction of hydrogen peroxide and organic hydroperoxides to water and alcohols, respectively. Plays a role in cell protection against oxidative stress by detoxifying peroxides and as sensor of hydrogen peroxide-mediated signaling events. Might participate in the signaling cascades of growth factors and tumor necrosis factor-alpha by regulating the intracellular concentrations of H(2)O(2) (PubMed:9497357). Reduces an intramolecular disulfide bond in G
CytoplasmMelanosome
Essential component of the TIM23 complex, a complex that mediates the translocation of transit peptide-containing proteins across the mitochondrial inner membrane (PubMed:30190335, PubMed:38828998). Has some phosphatase activity in vitro; however such activity may not be relevant in vivo May participate in the release of snRNPs and SMN from the Cajal body
Mitochondrion inner membraneNucleus speckle
3-methylglutaconic aciduria 9
An autosomal recessive disease characterized by early-onset seizures, severely delayed psychomotor development and intellectual disability. Patients have hypotonia or spasticity, and laboratory investigations show increased serum lactate and 3-methylglutaconic aciduria.
Acyltransferase required to remodel newly synthesized phospholipid cardiolipin (1',3'-bis-[1,2-diacyl-sn-glycero-3-phospho]-glycerol or CL), a key component of the mitochondrial inner membrane, with tissue specific acyl chains necessary for adequate mitochondrial function (PubMed:12930833, PubMed:19164547, PubMed:19700766, PubMed:26908608, PubMed:33096711). Its role in cellular physiology is to improve mitochondrial performance (PubMed:32234310). CL is critical for the coassembly of lipids and p
Mitochondrion outer membraneMitochondrion inner membraneMitochondrion membraneCytoplasm
Barth syndrome
An X-linked disease characterized by dilated cardiomyopathy with endocardial fibroelastosis, a predominantly proximal skeletal myopathy, growth retardation, neutropenia, and organic aciduria, particularly excess of 3-methylglutaconic acid. Additional features include hypertrophic cardiomyopathy, isolated left ventricular non-compaction, ventricular arrhythmia, motor delay, poor appetite, fatigue and exercise intolerance, hypoglycemia, lactic acidosis, hyperammonemia, and dramatic late catch-up growth after growth delay throughout childhood.
Component of the MICOS complex, a large protein complex of the mitochondrial inner membrane that plays crucial roles in the maintenance of crista junctions, inner membrane architecture, and formation of contact sites to the outer membrane (PubMed:25997101, PubMed:27623147, PubMed:32567732). Constituent of mature MICOS complex, it is required for the formation of cristae junction (CJ) and maintenance of cristae morphology (PubMed:25997101, PubMed:27623147, PubMed:32567732). Required for the incor
Mitochondrion inner membrane
Combined oxidative phosphorylation deficiency 37
An autosomal recessive disorder due to mitochondrial dysfunction and characterized by hypotonia, failure to thrive, progressive neurodegeneration with neurologic deterioration after the first months of life, global developmental delay, as well as liver dysfunction. Some patients may have hypertrophic cardiomyopathy, loss of vision and hearing, and/or seizures. Death in first months or years of life is observed in most patients.
Catalyzes the initial reaction in intramitochondrial fatty acid synthesis, by activating malonate and methylmalonate, but not acetate, into their respective CoA thioester (PubMed:21841779, PubMed:21846720). May have some preference toward very-long-chain substrates (PubMed:17762044)
Mitochondrion
Combined malonic and methylmalonic aciduria
A metabolic disease characterized by malonic and methylmalonic aciduria, with urinary excretion of much larger amounts of methylmalonic acid than malonic acid, in the presence of normal malonyl-CoA decarboxylase activity. Clinical features include coma, ketoacidosis, hypoglycemia, failure to thrive, microcephaly, dystonia, axial hypotonia and/or developmental delay, and neurologic manifestations including seizures, psychiatric disease and/or cognitive decline.
Transcription factor, which has both transcriptional activation and repression activities (PubMed:31905202). Also modulates chromatin accessibility (PubMed:38361031). In complex with HCFC1 and ZNF143, regulates the expression of several genes, including AP2S1, ESCO2, OPHN1, RBL1, UBXN8 and ZNF32 (PubMed:26416877). May regulate the expression of genes that encode both cytoplasmic and mitochondrial ribosomal proteins (By similarity). Required for normal mitochondrial development and function. Regu
NucleusCytoplasm
Methylmalonic aciduria and homocystinuria type cblL
An autosomal recessive disorder of cobalamin metabolism clinically characterized by early-onset seizures, and profound global developmental delay with severe intellectual disability. Metabolic features are mild methylmalonic aciduria, low-normal plasma methionine, and high-normal plasma homocysteine.
Serine/threonine-protein phosphatase component of macronutrients metabolism. Forms a functional kinase and phosphatase pair with BCKDK, serving as a metabolic regulatory node that coordinates branched-chain amino acids (BCAAs) with glucose and lipid metabolism via two distinct phosphoprotein targets: mitochondrial BCKDHA subunit of the branched-chain alpha-ketoacid dehydrogenase (BCKDH) complex and cytosolic ACLY, a lipogenic enzyme of Krebs cycle (PubMed:17336929, PubMed:17374715, PubMed:194117
Mitochondrion matrix
Maple syrup urine disease, mild variant
A mild form of maple syrup urine disease, a metabolic disorder due to an enzyme defect in the catabolic pathway of the branched-chain amino acids leucine, isoleucine, and valine. Accumulation of these 3 amino acids and their corresponding keto acids leads to encephalopathy and progressive neurodegeneration. Clinical features include mental and physical retardation, feeding problems, and a maple syrup odor to the urine. The keto acids of the branched-chain amino acids are present in the urine. If untreated, maple syrup urine disease can lead to seizures, coma, and death. The disease is often classified by its pattern of signs and symptoms. The most common and severe form of the disease is the classic type, which becomes apparent soon after birth. Variant forms of the disorder become apparent later in infancy or childhood and are typically milder, but they still involve developmental delay and other medical problems if not treated. MSUDMV is characterized by increased plasma levels of branched-chain amino acids (BCAA) apparent at birth. Treatment with a low-protein diet free of BCAA can result in normal psychomotor development and lack of metabolic episodes.
Catalytic release of biotin from biocytin, the product of biotin-dependent carboxylases degradation
Secreted, extracellular space
Biotinidase deficiency
A juvenile form of multiple carboxylase deficiency, an autosomal recessive disorder of biotin metabolism, characterized by ketoacidosis, hyperammonemia, excretion of abnormal organic acid metabolites, and dermatitis. Biotinidase deficiency is characterized by seizures, hypotonia, skin rash, alopecia, ataxia, hearing loss, and optic atrophy. If untreated, symptoms usually become progressively worse, and coma and death may occur.
Lysosomal membrane chaperone required to export cobalamin (vitamin B12) from the lysosome to the cytosol, allowing its conversion to cofactors (PubMed:19136951). Targets ABCD4 transporter from the endoplasmic reticulum to the lysosome (PubMed:27456980). Then forms a complex with lysosomal ABCD4 and cytoplasmic MMACHC to transport cobalamin across the lysosomal membrane (PubMed:25535791). Acts as an adapter protein which plays an important role in mediating and regulating the internalization of t
Endoplasmic reticulum membraneLysosome membraneCell membraneCytoplasmic vesicle, clathrin-coated vesicle
Methylmalonic aciduria and homocystinuria, cblF type
An autosomal recessive disorder of cobalamin metabolism characterized by decreased levels of the coenzymes adenosylcobalamin (AdoCbl) and methylcobalamin (MeCbl). It is due to accumulation of free cobalamin in lysosomes, thus hindering its conversion to cofactors. Clinical features include developmental delay, stomatitis, glossitis, seizures and methylmalonic aciduria responsive to vitamin B12.
Catalyzes the reversible isomerization of methylmalonyl-CoA (MMCoA) (generated from branched-chain amino acid metabolism and degradation of dietary odd chain fatty acids and cholesterol) to succinyl-CoA (3-carboxypropionyl-CoA), a key intermediate of the tricarboxylic acid cycle
Mitochondrion matrixMitochondrionCytoplasm
Methylmalonic aciduria due to methylmalonyl-CoA mutase deficiency
An often fatal disorder of organic acid metabolism. Common clinical features include lethargy, vomiting, failure to thrive, hypotonia, neurological deficit and early death. Two forms of the disease are distinguished by the presence (mut-) or absence (mut0) of residual enzyme activity. Mut0 patients have more severe neurological manifestations of the disease than do MUT- patients. MAMM is unresponsive to vitamin B12 therapy.
Carboxyltransferase subunit of the 3-methylcrotonyl-CoA carboxylase, an enzyme that catalyzes the conversion of 3-methylcrotonyl-CoA to 3-methylglutaconyl-CoA, a critical step for leucine and isovaleric acid catabolism
Mitochondrion matrix
3-methylcrotonoyl-CoA carboxylase 2 deficiency
An autosomal recessive disorder of leucine catabolism. The phenotype is variable, ranging from neonatal onset with severe neurological involvement to asymptomatic adults. There is a characteristic organic aciduria with massive excretion of 3-hydroxyisovaleric acid and 3-methylcrotonylglycine, usually in combination with a severe secondary carnitine deficiency.
Involved in cobalamin metabolism and trafficking (PubMed:18385497, PubMed:23415655, PubMed:24722857, PubMed:26364851). Plays a role in regulating the biosynthesis and the proportion of two coenzymes, methylcob(III)alamin (MeCbl) and 5'-deoxyadenosylcobalamin (AdoCbl) (PubMed:18385497, PubMed:23415655, PubMed:24722857). Promotes oxidation of cob(II)alamin bound to MMACHC (PubMed:26364851). The processing of cobalamin in the cytosol occurs in a multiprotein complex composed of at least MMACHC, MMA
CytoplasmMitochondrion
Methylmalonic aciduria and homocystinuria, cblD type
An autosomal recessive disorder of cobalamin metabolism characterized by decreased levels of the coenzymes adenosylcobalamin (AdoCbl) and methylcobalamin (MeCbl). Clinical features include developmental delay, hyotonia, intellectual disability, seizures, and megaloblastic anemia. Laboratory studies show methylmalonic aciduria and homocystinuria.
Mitochondrial co-chaperone which forms a complex with prohibitins to regulate cardiolipin remodeling (By similarity). May be a component of the PAM complex, a complex required for the translocation of transit peptide-containing proteins from the inner membrane into the mitochondrial matrix in an ATP-dependent manner. May act as a co-chaperone that stimulate the ATP-dependent activity (By similarity)
Mitochondrion inner membrane
3-methylglutaconic aciduria 5
An autosomal recessive disorder characterized by early-onset dilated cardiomyopathy, growth failure, cerebellar ataxia causing significant motor delays, testicular dysgenesis, growth failure and significant increases in urine organic acids, particularly 3-methylglutaconic acid and 3-methylglutaric acid.
Together with BCKDHA forms the heterotetrameric E1 subunit of the mitochondrial branched-chain alpha-ketoacid dehydrogenase (BCKD) complex. The BCKD complex catalyzes the multi-step oxidative decarboxylation of alpha-ketoacids derived from the branched-chain amino-acids valine, leucine and isoleucine producing CO2 and acyl-CoA which is subsequently utilized to produce energy. The E1 subunit catalyzes the first step with the decarboxylation of the alpha-ketoacid forming an enzyme-product intermed
Mitochondrion matrix
Maple syrup urine disease 1B
A form of maple syrup urine disease, an autosomal recessive metabolic disorder due to an enzyme defect in the catabolic pathway of the branched-chain amino acids leucine, isoleucine, and valine. Accumulation of these 3 amino acids and their corresponding keto acids leads to encephalopathy and progressive neurodegeneration. Clinical features include mental and physical retardation, feeding problems, and a maple syrup odor to the urine. The keto acids of the branched-chain amino acids are present in the urine. If untreated, maple syrup urine disease can lead to seizures, coma, and death. The disease is often classified by its pattern of signs and symptoms. The most common and severe form of the disease is the classic type, which becomes apparent soon after birth. Variant forms of the disorder become apparent later in infancy or childhood and are typically milder, but they still involve developmental delay and other medical problems if not treated.
Facilitates the transport of serine from the cytosol to the mitochondria by interacting with and stabilizing Sideroflexin-1 (SFXN1), a mitochondrial serine transporter, playing a fundamental role in the one-carbon cycle responsible for the synthesis of nucleotides needed for mitochondrial DNA replication (PubMed:35235340). Plays an important role in the phosphatidylglycerol (PG) remodeling that is essential for both mitochondrial function and intracellular cholesterol trafficking (PubMed:2268371
Mitochondrion membraneEndoplasmic reticulumMitochondrion
3-methylglutaconic aciduria with deafness, encephalopathy, and Leigh-like syndrome
An autosomal recessive disorder characterized by childhood onset of delayed psychomotor development or psychomotor regression, sensorineural deafness, spasticity or dystonia, and increased excretion of 3-methylglutaconic acid. Brain imaging shows cerebral and cerebellar atrophy as well as lesions in the basal ganglia reminiscent of Leigh syndrome. Laboratory studies show increased serum lactate and alanine, mitochondrial oxidative phosphorylation defects, abnormal mitochondria, abnormal phosphatidylglycerol and cardiolipin profiles in fibroblasts, and abnormal accumulation of unesterified cholesterol within cells.
Hydrolyzes 3-hydroxyisobutyryl-CoA (HIBYL-CoA), a saline catabolite. Has high activity toward isobutyryl-CoA. Could be an isobutyryl-CoA dehydrogenase that functions in valine catabolism. Also hydrolyzes 3-hydroxypropanoyl-CoA
Mitochondrion
3-hydroxyisobutryl-CoA hydrolase deficiency
An autosomal recessive inborn error of valine metabolism. It causes severely delayed psychomotor development, neurodegeneration, increased lactic acid, and brain lesions in the basal ganglia.
Catalyzes the formation of fatty acid-cholesterol esters, which are less soluble in membranes than cholesterol (PubMed:16154994, PubMed:16647063, PubMed:32433613, PubMed:32433614, PubMed:32944968, PubMed:9020103). Plays a role in lipoprotein assembly and dietary cholesterol absorption (PubMed:16154994, PubMed:9020103). Preferentially utilizes oleoyl-CoA ((9Z)-octadecenoyl-CoA) as a substrate: shows a higher activity towards an acyl-CoA substrate with a double bond at the delta-9 position (9Z) th
Endoplasmic reticulum membrane
Serine protease that shows proteolytic activity against a non-specific substrate beta-casein (PubMed:10873535). Promotes apoptosis by either relieving the inhibition of BIRC proteins on caspases, leading to an increase in caspase activity; or by a BIRC inhibition-independent, caspase-independent and serine protease activity-dependent mechanism (PubMed:15200957). Cleaves BIRC6 and relieves its inhibition on CASP3, CASP7 and CASP9, but it is also prone to inhibition by BIRC6 (PubMed:36758104, PubM
Mitochondrion intermembrane spaceMitochondrion membraneEndoplasmic reticulum
3-methylglutaconic aciduria 8
An autosomal recessive inborn error of metabolism resulting in early death. Clinical features include extreme hypertonia observed at birth, alternating with hypotonia, subsequent appearance of extrapyramidal symptoms, lack of psychomotor development, microcephaly, and intractable seizures. Patients show lactic acidemia, 3-methylglutaconic aciduria, intermittent neutropenia, and progressive brain atrophy.
Catalyzes the conversion of malonyl-CoA to acetyl-CoA. In the fatty acid biosynthesis MCD selectively removes malonyl-CoA and thus assures that methyl-malonyl-CoA is the only chain elongating substrate for fatty acid synthase and that fatty acids with multiple methyl side chains are produced. In peroxisomes it may be involved in degrading intraperoxisomal malonyl-CoA, which is generated by the peroxisomal beta-oxidation of odd chain-length dicarboxylic fatty acids. Plays a role in the metabolic
CytoplasmMitochondrion matrixPeroxisomePeroxisome matrix
Malonyl-CoA decarboxylase deficiency
Autosomal recessive disease characterized by abdominal pain, chronic constipation, episodic vomiting, metabolic acidosis and malonic aciduria.
Catalyzes the oxidative decarboxylation of glutaryl-CoA to crotonyl-CoA and CO(2) in the degradative pathway of L-lysine, L-hydroxylysine, and L-tryptophan metabolism. It uses electron transfer flavoprotein as its electron acceptor. Isoform Short is inactive
Mitochondrion matrix
Glutaric aciduria 1
An autosomal recessive metabolic disorder characterized by progressive dystonia and athetosis due to gliosis and neuronal loss in the basal ganglia.
A mitochondrial matrix enzyme that catalyzes the third step in leucine catabolism, where isovaleryl-CoA (3-methylbutanoyl-CoA) is metabolized to 3-methylbut-2-enoyl-CoA (PubMed:7640268). To a lesser extent, it also participates in the first step in fatty acid beta-oxidation, in which it catalyzes the proR-proR stereospecific alpha,beta-dehydrogenation of other saturated short-chain acyl-CoA thioesters such as pentanoyl-CoA, hexanoyl-CoA and butanoyl-CoA, using the electron transfer flavoprotein
Mitochondrion matrix
Isovaleric acidemia
A metabolic disorder characterized by retarded psychomotor development, a peculiar odor resembling sweaty feet, an aversion to dietary protein, and pernicious vomiting, leading to acidosis and coma. The acute neonatal form leads to massive metabolic acidosis from the first days of life and rapid death.
Converts cob(I)alamin to adenosylcobalamin (adenosylcob(III)alamin), a coenzyme for methylmalonyl-CoA mutase, therefore participates in the final step of the vitamin B12 conversion (PubMed:12514191). Generates adenosylcobalamin (AdoCbl) and directly delivers the cofactor to MUT in a transfer that is stimulated by ATP-binding to MMAB and gated by MMAA (Probable)
Mitochondrion
Methylmalonic aciduria, cblB type
An autosomal recessive disorder of methylmalonate and cobalamin metabolism due to defective synthesis of adenosylcobalamin.
Receptor for transcobalamin saturated with cobalamin (TCbl) (PubMed:18779389). Plays an important role in cobalamin uptake (PubMed:18779389, PubMed:20524213). Plasma membrane protein that is expressed on follicular dendritic cells (FDC) and mediates interaction with germinal center B cells (PubMed:10727470). Functions as costimulator to promote B cell responses to antigenic stimuli; promotes B cell differentiation and proliferation (PubMed:10727470, PubMed:11418631). Germinal center-B (GC-B) cel
Cell membrane
Methylmalonic aciduria, transient, due to transcobalamin receptor defect
An autosomal recessive metabolic condition characterized by moderate methymalonicaciduria, and normal plasma vitamin B12 levels. Serum homocysteine may be increased in some affected individuals. Most cases are clinically asymptomatic.
Together with BCKDHB forms the heterotetrameric E1 subunit of the mitochondrial branched-chain alpha-ketoacid dehydrogenase (BCKD) complex. The BCKD complex catalyzes the multi-step oxidative decarboxylation of alpha-ketoacids derived from the branched-chain amino-acids valine, leucine and isoleucine producing CO2 and acyl-CoA which is subsequently utilized to produce energy. The E1 subunit catalyzes the first step with the decarboxylation of the alpha-ketoacid forming an enzyme-product intermed
Mitochondrion matrix
Maple syrup urine disease 1A
A form of maple syrup urine disease, an autosomal recessive metabolic disorder due to an enzyme defect in the catabolic pathway of the branched-chain amino acids leucine, isoleucine, and valine. Accumulation of these 3 amino acids and their corresponding keto acids leads to encephalopathy and progressive neurodegeneration. Clinical features include mental and physical retardation, feeding problems, and a maple syrup odor to the urine. The keto acids of the branched-chain amino acids are present in the urine. If untreated, maple syrup urine disease can lead to seizures, coma, and death. The disease is often classified by its pattern of signs and symptoms. The most common and severe form of the disease is the classic type, which becomes apparent soon after birth. Variant forms of the disorder become apparent later in infancy or childhood and are typically milder, but they still involve developmental delay and other medical problems if not treated.
Coenzyme A (CoA) transferase that reversibly catalyzes the transfer of a CoA moiety from a dicarboxyl-CoA to a dicarboxylate in a metabolite recycling process. Displays preference for succinyl-CoA and glutarate-CoA as dicarboxyl-CoA donors and glutarate, succinate, adipate/hexanedioate, itaconate and 3-hydroxy-3-methylglutarate as dicarboxylate acceptors (PubMed:23893049, PubMed:34492704, PubMed:38915184). Acts on intermediates or end products of lysine and tryptophan degradation pathway, in par
Mitochondrion
Glutaric aciduria 3
An autosomal recessive metabolic condition characterized by urinary excretion of abnormal quantities of glutaric acid, in the presence of normal 3-hydroxyglutarate, glutarylcarnitine and glutarylglycine urinary levels. Affected individuals show no consistent clinical phenotype and many are asymptomatic.
Lysosomal membrane protein that transports cobalamin (Vitamin B12) from the lysosomal lumen to the cytosol in an ATP-dependent manner (PubMed:22922874, PubMed:28572511, PubMed:31467407, PubMed:33845046). Targeted by LMBRD1 lysosomal chaperone from the endoplasmic reticulum to the lysosomal membrane (PubMed:27456980). Then forms a complex with lysosomal chaperone LMBRD1 and cytosolic MMACHC to transport cobalamin across the lysosomal membrane (PubMed:25535791)
Endoplasmic reticulum membraneLysosome membrane
Methylmalonic aciduria and homocystinuria type cblJ
A disorder of cobalamin metabolism characterized by decreased levels of the coenzymes adenosylcobalamin (AdoCbl) and methylcobalamin (MeCbl). Clinical features include feeding difficulties, poor growth, hypotonia, lethargy, anemia, and developmental delay.
Lipoamide dehydrogenase is a component of the glycine cleavage system as well as an E3 component of three alpha-ketoacid dehydrogenase complexes (pyruvate-, alpha-ketoglutarate-, and branched-chain amino acid-dehydrogenase complex) (PubMed:15712224, PubMed:16442803, PubMed:16770810, PubMed:17404228, PubMed:20160912, PubMed:20385101). The 2-oxoglutarate dehydrogenase complex is mainly active in the mitochondrion (PubMed:29211711). A fraction of the 2-oxoglutarate dehydrogenase complex also locali
Mitochondrion matrixNucleusCell projection, cilium, flagellumCytoplasmic vesicle, secretory vesicle, acrosome
Dihydrolipoamide dehydrogenase deficiency
An autosomal recessive metabolic disorder characterized biochemically by a combined deficiency of the branched-chain alpha-keto acid dehydrogenase complex (BCKDC), pyruvate dehydrogenase complex (PDC), and alpha-ketoglutarate dehydrogenase complex (KGDC). Clinically, affected individuals have lactic acidosis and neurologic deterioration due to sensitivity of the central nervous system to defects in oxidative metabolism.
Functions as a regulatory ATPase and participates in secretion/protein trafficking process. Has ATP-dependent protein disaggregase activity and is required to maintain the solubility of key mitochondrial proteins (PubMed:32573439, PubMed:34115842, PubMed:35247700, PubMed:36170828, PubMed:36745679). Involved in mitochondrial-mediated antiviral innate immunity, activates RIG-I-mediated signal transduction and production of IFNB1 and pro-inflammatory cytokine IL6 (PubMed:31522117). Plays a role in
Mitochondrion intermembrane space
3-methylglutaconic aciduria 7B
An autosomal recessive inborn error of metabolism with a highly variable phenotype. Primary disease symptoms are increased levels of 3-methylglutaconic acid, neurologic deterioration and neutropenia. Other common features include progressive encephalopathy, movement abnormalities, delayed psychomotor development,impaired intellectual development, cataracts, seizures, and recurrent infections.
Methylmalonyl-CoA epimerase involved in propionyl-CoA metabolism
Mitochondrion
Methylmalonyl-CoA epimerase deficiency
Autosomal recessive inborn error of amino acid metabolism, involving valine, threonine, isoleucine and methionine. This organic aciduria may present in the neonatal period with life-threatening metabolic acidosis, hyperammonemia, feeding difficulties, pancytopenia and coma.
Isobutyryl-CoA dehydrogenase which catalyzes the conversion of 2-methylpropanoyl-CoA to (2E)-2-methylpropenoyl-CoA in the valine catabolic pathway (PubMed:11013134, PubMed:12359132, PubMed:16857760). To a lesser extent, also able to catalyze the oxidation of (2S)-2-methylbutanoyl-CoA (PubMed:11013134, PubMed:12359132)
Mitochondrion
Isobutyryl-CoA dehydrogenase deficiency
An autosomal recessive metabolic disorder characterized by plasma carnitine deficiency and elevated C4-acylcarnitine. Patients manifest variable clinical features including failure to thrive, seizures, anemia, muscular hypotonia and developmental delay. Some patients may be asymptomatic.
Short and branched chain specific acyl-CoA dehydrogenase that catalyzes the proR-proR stereospecific alpha,beta-dehydrogenation of fatty acyl-CoA thioesters using the electron transfer flavoprotein (ETF) as their physiologic electron acceptor, resulting in the formation of trans-2-enoyl-CoA ((2E)-enoyl-CoA) (PubMed:10832746, PubMed:11013134, PubMed:21430231, PubMed:7698750). Among the different mitochondrial acyl-CoA dehydrogenases, acts specifically on short and branched chain acyl-CoA derivati
Mitochondrion matrix
Short/branched-chain acyl-CoA dehydrogenase deficiency
Autosomal recessive disorder and consists of a defect in catabolism of L-isoleucine which is characterized by an increase of 2-methylbutyrylglycine and 2-methylbutyrylcarnitine in blood and urine. Affected individuals have seizures and psychomotor delay as the main clinical features.
Cobalamin (vitamin B12) cytosolic chaperone that catalyzes the reductive decyanation of cyanocob(III)alamin (cyanocobalamin, CNCbl) to yield cob(II)alamin and cyanide, using FAD or FMN as cofactors and NADPH as cosubstrate (PubMed:18779575, PubMed:19700356, PubMed:21697092, PubMed:25809485). Cyanocobalamin constitutes the inactive form of vitamin B12 introduced from the diet, and is converted into the active cofactors methylcobalamin (MeCbl) involved in methionine biosynthesis, and 5'-deoxyadeno
Cytoplasm, cytosol
Methylmalonic aciduria and homocystinuria, cblC type
An autosomal recessive disorder of cobalamin metabolism characterized by decreased levels of the coenzymes adenosylcobalamin (AdoCbl) and methylcobalamin (MeCbl). Affected individuals may have developmental, hematologic, neurologic, metabolic, ophthalmologic, and dermatologic clinical findings. Although considered a disease of infancy or childhood, some individuals develop symptoms in adulthood.
Transcriptional coregulator (By similarity). Serves as a scaffold protein, bridging interactions between transcription factors, including THAP11 and ZNF143, and transcriptional coregulators (PubMed:26416877). Involved in control of the cell cycle (PubMed:10629049, PubMed:10779346, PubMed:15190068, PubMed:16624878, PubMed:23629655). Also antagonizes transactivation by ZBTB17 and GABP2; represses ZBTB17 activation of the p15(INK4b) promoter and inhibits its ability to recruit p300 (PubMed:10675337
CytoplasmNucleus
Methylmalonic aciduria and homocystinuria, cblX type
An X-linked recessive metabolic disorder characterized by severely delayed psychomotor development apparent in infancy, failure to thrive, impaired intellectual development, and intractable epilepsy. Additional features may include microcephaly and choreoathetosis.
Malonate and methylmalonate semialdehyde dehydrogenase involved in the catabolism of valine, thymine, and compounds catabolized by way of beta-alanine, including uracil and cytidine
Mitochondrion
Methylmalonate semialdehyde dehydrogenase deficiency
A metabolic disorder characterized by elevated beta-alanine, 3-hydroxypropionic acid, and both isomers of 3-amino and 3-hydroxyisobutyric acids in urine organic acids.
Biotin--protein ligase catalyzing the biotinylation of the 4 biotin-dependent carboxylases acetyl-CoA-carboxylase, pyruvate carboxylase, propionyl-CoA carboxylase, and methylcrotonyl-CoA carboxylase
CytoplasmMitochondrion
Holocarboxylase synthetase deficiency
A neonatal form of multiple carboxylase deficiency, an autosomal recessive disorder of biotin metabolism, characterized by ketoacidosis, hyperammonemia, excretion of abnormal organic acid metabolites, and dermatitis. In holocarboxylase synthetase deficiency, clinical and biochemical symptoms improve dramatically with administration of biotin.
This is one of the 2 subunits of the biotin-dependent propionyl-CoA carboxylase (PCC), a mitochondrial enzyme involved in the catabolism of odd chain fatty acids, branched-chain amino acids isoleucine, threonine, methionine, and valine and other metabolites (PubMed:6765947, PubMed:8434582). Propionyl-CoA carboxylase catalyzes the carboxylation of propionyl-CoA/propanoyl-CoA to D-methylmalonyl-CoA/(S)-methylmalonyl-CoA (PubMed:10101253, PubMed:6765947, PubMed:8434582). Within the holoenzyme, the
Mitochondrion matrix
Propionic acidemia type I
Life-threatening disease characterized by episodic vomiting, lethargy and ketosis, neutropenia, periodic thrombocytopenia, hypogammaglobulinemia, developmental retardation, and intolerance to protein.
Medicamentos e terapias
Mecanismo: Carbamoyl-phosphate synthase [ammonia], mitochondrial positive allosteric modulator
Variantes genéticas (ClinVar)
533 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
53 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Acidúria orgânica
Centros de Referência SUS
21 centros habilitados pelo SUS para Acidúria orgânica
Centros para Acidúria orgânica
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
NUPAD / Faculdade de Medicina UFMG
Av. Prof. Alfredo Balena, 189 - 5 andar - Centro, Belo Horizonte - MG, 30130-100 · CNES 2183226
Serviço de Referência
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital de Clínicas da Universidade Federal de Pernambuco
Av. Prof. Moraes Rego, 1235 - Cidade Universitária, Recife - PE, 50670-901 · CNES 2561492
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital Universitário Onofre Lopes (HUOL)
Av. Nilo Peçanha, 620 - Petrópolis, Natal - RN, 59012-300 · CNES 2408570
Atenção Especializada
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
Instituto da Criança e do Adolescente (ICr-HCFMUSP)
Av. Dr. Enéas Carvalho de Aguiar, 647 - Cerqueira César, São Paulo - SP, 05403-000 · CNES 2081695
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
3 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Brain morphometry and cognition in late-onset glutaric aciduria type 1: scoping review and novel insights from a case report.
Glutaric Aciduria type I (GA1) is a rare autosomal recessive organic aciduria, with typical early-onset presentation, characterized by severe movement disorders with damage to the basal ganglia following an acute encephalopathic crisis. Late-onset (LO), milder forms have rarely been described. In LO patients, severe brain damage is frequently reported, including frontotemporal hypoplasia, brain atrophy, subependymal nodules and leukodystrophy. Cognitive impairment is sometimes described, often without formal psychometric assessment. This scoping review aims to synthesize the neuroradiological and neuropsychological features of all LO GA1 patients reported in the literature to date, divided in undiagnosed LO cases detected through selective screening and LO cases with onset of symptoms after 6 years of age. The following databases were used: PubMed, Embase and Medline. The search strategy abides by the PRISMA-ScR guidelines. Out of the 53 LO patients reviewed, extensive quantitative neuropsychological testing was reported in seven cases, while no brain morphometric analysis was performed. CASE REPORT. A novel case of a GA1 34-year-old Chinese woman identified through the neonatal screening of her healthy baby is described. Brain morphometric analysis showed diffused reduced volumes and cortical thinning, involving fronto-temporal areas and, to a lesser extent, the parieto-occipital regions. The neuropsychological assessment highlighted mild difficulties in verbal executive functions (inferential thinking) and phonological short-term memory. The present review and case report suggest that integrating neuroanatomical and neuropsychological investigations into clinical practice may allow a more refined characterization of GA1 patients, contributing to unveil the complex ethio-pathogenic mechanisms underlying the disease and monitor patients over time. The online version contains supplementary material available at 10.1007/s10072-026-08886-9.
Nutritional Management in Severe Methylmalonic and Propionic Acidemias: How Much Medical Food Is Too Much?
Methylmalonic acidemia (MMA) and propionic acidemia (PA) are inherited metabolic disorders affecting valine and isoleucine catabolism. Long-term therapy mainly involves dietary protein restriction. An amino acid mixture (AAM, medical food) free of the precursor amino acids is frequently used, especially when protein intake does not reach World Health Organization (WHO) recommendations. However, its clinical impact on disease control and patient outcomes remains unclear. Our study aimed to retrospectively review the dietary prescriptions in a cohort of vitamin B12-unresponsive MMA and PA patients and to analyze their impact on clinical and laboratory parameters. Clinical data, anthropometric measurements and dietary prescriptions were collected from the patients' medical and dietary files. We included 71 patients (38 MMA and 33 PA). Fifty-nine percent of the patients' dietary prescriptions did not reach the safe WHO-recommended daily total protein intake. Among these, 28% included AAM supplementation versus 62% in the group of patients that met the WHO recommendations (p < 0.001). AAM was associated with a decrease in mean plasma concentrations of isoleucine and valine. These plasma amino acid concentrations were corrected by isoleucine and valine supplementation; however, leucine/isoleucine and leucine/valine ratios remained elevated in comparison to patients without AAM. Nutritional and clinical scores were worsened by AAM supplementation. We found that MMA/PA patients receiving AAM tend to have altered plasma amino acid concentrations, raising concerns about potential long-term deleterious consequences of AAM. We recommend prioritizing natural protein intake over AAM when possible, and if not, to carefully monitor and moderately supplement valine and isoleucine to prevent deficiencies. Oculocerebrorenal syndrome, more commonly referred to as Lowe syndrome, was first described in the early 1950s by Lowe and colleagues, who identified a constellation of findings in affected children, including intellectual disability, organic aciduria, reduced renal ammonia production, bilateral cataracts, and glaucoma. Subsequent studies have further delineated the disorder, noting the presence of Fanconi-type proximal renal tubulopathy, generalized areflexia, and characteristic arthropathy. The condition is now established as an X-linked recessive disorder caused by mutations in the OCRL gene, leading to disturbances in phosphoinositide metabolism and widespread impairment of intracellular trafficking and cytoskeletal organization. These molecular derangements explain the syndrome’s broad involvement of ocular, neurological, and renal systems. The syndrome primarily affects the lens and trabecular meshwork in the eye, resulting in congenital cataracts and infantile-onset glaucoma. In the central nervous system, abnormalities in synaptic signaling and white matter integrity underlie the intellectual disability, hypotonia, and seizures observed in many patients. Renal manifestations arise from dysfunction of the proximal tubules, producing a partial Fanconi syndrome characterized by bicarbonaturia, phosphaturia, aminoaciduria, and progressive chronic kidney disease. The natural history of oculocerebrorenal syndrome typically begins in the neonatal period, characterized by severe muscular hypotonia and ocular abnormalities, which serve as the earliest diagnostic indicators. Renal manifestations generally evolve more gradually, with incomplete Fanconi syndrome becoming evident in infancy or early childhood. Over time, the progression of renal disease culminates in chronic kidney failure, which remains the leading cause of mortality. Life expectancy is limited, with most patients surviving into the third or fourth decade of life. Early diagnosis and supportive care are crucial for preventing life-threatening complications and maximizing the quality of life.
Global Longitudinal Strain Alteration of the Left Ventricle in Children with Organic Aciduria: Cardiac Disease in Organic Aciduria.
Introduction: Cardiac complications are well-documented in propionic acidemia (PA), and there are a few reported cases of cardiomyopathies in methylmalonic acidemia (MMA). Left-ventricular global longitudinal strain (LV GLS) measurement is known to be able to detect early ventricular dysfunction, leading potentially to cardiomyopathy. The aim of our study was to evaluate left-ventricular global longitudinal strain (LV GLS) in MMA and PA patients and compare it with the pediatric general population. Methods: In this monocentric retrospective study, 26 patients with organic aciduria (OA) were included. Demographic, clinical, electrocardiographic and echocardiographic data were collected. The mean LV GLS in MMA and PA patients was compared with the GLS in the pediatric general population. Results: The left-ventricular ejection fraction (LVEF) was similar between MMA and PA patients and in the normal range (66.27 ± 6.24% vs. 61.41 ± 11.02%; p = 0.182). LV GLS was significantly lower in PA patients than in MMA patients (-15.8 ± 5.67% vs. -20.6 ± 3.19%; p = 0.011). LV GLS was significantly lower in PA patients when compared with the general pediatric population (p = 0.029). Conclusions: Patients with propionic acidemia may have impaired global longitudinal strain even in the presence of normal LVEF. LV GLS might be a useful tool for cardiac follow-up in pediatric patients with OA.
The Association Between Metabolomic and Usual Biochemical Data Helps to Detect Insulin Resistance.
Background: Chronic noncommunicable diseases account for nearly 80% of global deaths and are strongly associated with insulin resistance (IR). One of the most significant clinical findings of the past two decades is that the molecular mechanisms underlying immune and metabolic systems have been evolutionarily conserved across species. Methods: This study included 34 volunteers (19 men and 15 women). Demographic data were collected using validated questionnaires. Anthropometric measurements (weight, height, waist-to-hip ratio, and body composition assessed by tetrapolar bioimpedance) were obtained directly. Laboratory analyses included fasting glucose and insulin, glycated hemoglobin, HDL cholesterol, total cholesterol, triglycerides, organic aciduria, and additional biochemical markers assessed using standard methods. Group comparisons were performed using parametric or nonparametric statistical tests according to data distribution, as specified in the figure legends. Results: The primary analyses focused on identifying early metabolomic alterations associated with insulin resistance in individuals whose conventional biochemical parameters were within laboratory reference ranges. Individuals with a TG/HDL ratio > 2 and increased urinary kynurenate excretion exhibited a 3.6-fold higher relative risk of insulin resistance, while elevated insulin levels combined with urinary α-ketoisovalerate were associated with a 2.7-fold increased risk. Significant differences in plasma insulin, HbA1c, and HOMA-IR were observed between healthy and diseased individuals (p < 0.05), indicating early metabolic dysfunction preceding clinical disease onset. Conclusions: Metabolomic biomarkers serve as reliable indicators of subclinical metabolic disturbances, revealing significant risks in major metabolic pathways even in individuals with conventional exams within normal limits. Early detection through these metabolomic markers may enable personalized interventions aimed at preserving cellular function and systemic metabolic balance.
Clinical and neuroradiologic spectrum of glutaric acidemia type 1 in children: insights from a retrospective cohort in Guangdong Province, China.
Glutaric acidemia type 1 (GA-1) is a rare autosomal recessive metabolic disorder resulting from a deficiency in glutaryl-CoA dehydrogenase (GCDH). Current evidence indicates that GA-1 remains under-recognized by clinicians, a factor that may contribute to delayed diagnosis. The aim of this study was to retrospectively analyze the clinical manifestations and imaging characteristics of GA-1. This study enrolled patients diagnosed with GA-1 at the Guangzhou Women and Children's Medical Center between April 2014 and April 2024. Clinical data related to GA-1 were retrieved through the electronic medical record system, and magnetic resonance imaging (MRI) scans were collected for all patients. Cranial MRI images were independently evaluated by two radiologists (with 10 and 6 years of experience in pediatric neuroimaging diagnosis, respectively) using a blinded approach. Blood acylcarnitine levels were analyzed using tandem mass spectrometry, urinary organic acid concentrations were quantified via gas chromatography-mass spectrometry, and GCDH gene analysis was performed in a subset of patients. This study enrolled 24 GA-1 children (8 males, 16 females) from Guangdong Province, China. Diagnosis was confirmed by elevated glutaric acid (GA), 3-hydroxyglutaric acid (3-HGA), and glutarylcarnitine (C5DC) levels, with increased C5DC/octanoylcarnitine (C8) and C5DC/propionylcarnitine (C3) ratios. Genetic analysis identified 12 GCDH mutations in 11 patients, including 5 novel variants (c.395G>A, c.271+1G>A, c.1156C>G, c.146_149delACTG, and c.1011A>G). Neuroimaging revealed abnormal brain MRI findings in all patients (100%), predominantly featuring frontotemporal extracerebral space widening (75.0%, 18/24) and symmetric basal ganglia hyperintensity (83.3%, 20/24). These findings align with the established GA-1 phenotypes. This study underscores the need for heightened awareness of GA-1 among clinicians and radiologists, characterizes its MRI signature, and expands the GCDH mutation spectrum with five novel variants, thereby offering valuable guidance for imaging-based diagnosis and genetic counselling.
Publicações recentes
Brain morphometry and cognition in late-onset glutaric aciduria type 1: scoping review and novel insights from a case report.
Global Longitudinal Strain Alteration of the Left Ventricle in Children with Organic Aciduria: Cardiac Disease in Organic Aciduria.
🥉 Relato de casoThe Association Between Metabolomic and Usual Biochemical Data Helps to Detect Insulin Resistance.
Clinical and neuroradiologic spectrum of glutaric acidemia type 1 in children: insights from a retrospective cohort in Guangdong Province, China.
Whole exome sequencing in pediatric hyperammonemia: significant diagnostic yield and identification of three novel variants.
📚 EuropePMC54 artigos no totalmostrando 96
Brain morphometry and cognition in late-onset glutaric aciduria type 1: scoping review and novel insights from a case report.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyGlobal Longitudinal Strain Alteration of the Left Ventricle in Children with Organic Aciduria: Cardiac Disease in Organic Aciduria.
Journal of clinical medicineThe Association Between Metabolomic and Usual Biochemical Data Helps to Detect Insulin Resistance.
BiomedicinesClinical and neuroradiologic spectrum of glutaric acidemia type 1 in children: insights from a retrospective cohort in Guangdong Province, China.
Quantitative imaging in medicine and surgeryWhole exome sequencing in pediatric hyperammonemia: significant diagnostic yield and identification of three novel variants.
BMC medical genomicsNutritional Management in Severe Methylmalonic and Propionic Acidemias: How Much Medical Food Is Too Much?
Journal of inherited metabolic diseaseMitochondrial Acetoacetyl-CoA Thiolase Deficiency: Three New Cases Detected by Newborn Screening Confirming the Significance of C4OH Elevation.
International journal of neonatal screeningThe origin of abnormal organic acids in HMG-CoA synthase deficiency.
Molecular genetics and metabolism3-Hydroxy-3-methylglutaryl-coenzyme A lyase deficiency: case report of a child with rare HMGCL gene variants.
Journal of pediatric endocrinology & metabolism : JPEMThe broad spectrum of clinical manifestations observed in three patients with L2 hydroxyglutaric aciduria spans from febrile seizures to complex dystonia.
Molecular genetics and metabolism reportsGlutaric Aciduria Presenting With an Acute Encephalitic Crisis: A Case Report.
CureusCerebral White Matter Alterations Associated With Oligodendrocyte Vulnerability in Organic Acidurias: Insights in Glutaric Aciduria Type I.
Neurotoxicity researchThe cryo-EM structure of trypanosome 3-methylcrotonyl-CoA carboxylase provides mechanistic and dynamic insights into its enzymatic function.
Structure (London, England : 1993)Spectrum of Organic Aciduria Diseases in Tunisia: A 35-year Retrospective Study.
Saudi journal of medicine & medical sciencesPubertal origin of growth retardation in inborn errors of protein metabolism: A longitudinal cohort study.
Molecular genetics and metabolismFollow-up of Patients with Hereditary Metabolic Diseases during the 3 Years of the Pandemic in the Reference Center for Hereditary Metabolism Diseases of the Centro Hospitalar Universitário Lisboa Norte.
Endocrine, metabolic & immune disorders drug targetsCombined Oxidative Phosphorylation Deficiency Type-13 with Perinatal Presentation: A Case Report.
Endocrine, metabolic & immune disorders drug targetsClinical Presentation of Inherited Metabolic Diseases in Newborns Hospitalised in an Intensive Care Unit.
Journal of mother and childNovel pathogenic UQCRC2 variants in a female with normal neurodevelopment.
Cold Spring Harbor molecular case studiesGlutaryl-CoA Dehydrogenase Misfolding in Glutaric Acidemia Type 1.
International journal of molecular sciencesCase report: Variability in clinical features as a potential pitfall for the diagnosis of Barth syndrome.
Frontiers in pediatricsKidney urinary biomarkers in patients with branched-chain amino acid and cobalamin metabolism defects.
Journal of inherited metabolic diseaseDelayed Biotin Therapy in a Child with Atypical Profound Biotinidase Deficiency: Late Arrival of the Truth and a Lesson Worth Thinking.
International journal of molecular sciencesGlutaric aciduria and L-2-hydroxyglutaric aciduria: Clinical and molecular findings of 35 patients from Turkey.
Molecular genetics and metabolism reportsOrganic Aciduria Disorders in Pregnancy: An Overview of Metabolic Considerations.
Metabolites2-Methylglutaconic acid as a biomarker in routine urine organic acids leading to the diagnosis of glutaric acidemia type I in a low excretor.
Molecular genetics and metabolismHow guideline development has informed clinical research for organic acidurias (et vice versa).
Journal of inherited metabolic diseaseStroke-like Episodes in Inherited Neurometabolic Disorders.
MetabolitesScreening of Organic Acidurias by Gas Chromatography-Mass Spectrometry (GC-MS).
Methods in molecular biology (Clifton, N.J.)Cochlear Implantation in Biotinidase Enzyme Deficiency.
Indian journal of otolaryngology and head and neck surgery : official publication of the Association of Otolaryngologists of IndiaDiversion of Acetyl CoA to 3-Methylglutaconic Acid Caused by Discrete Inborn Errors of Metabolism.
MetabolitesExpert consensus on screening, diagnosis and treatment of multiple carboxylase deficiency.
Zhejiang da xue xue bao. Yi xue ban = Journal of Zhejiang University. Medical sciencesComparing amniotic fluid mass spectrometry assays and amniocyte gene analyses for the prenatal diagnosis of methylmalonic aciduria.
PloS oneIntracranial Calcification Associated with 3-Methylcrotonyl-CoA Carboxylase Deficiency.
Molecular syndromologyFunctional neurologic disorders in an adult with propionic acidemia: a case report.
BMC psychiatryOlivopontocerebellar degeneration associated with 3-hydroxy-3-methylglutaric aciduria in a domestic shorthair cat.
JFMS open reportsGlutaric Aciduria Type II With Ketosis in a Male Infant.
CureusThe first knock-in rat model for glutaric aciduria type I allows further insights into pathophysiology in brain and periphery.
Molecular genetics and metabolismParkinsonism and iron deposition in two adult patients with L-2-hydroxiglutaric aciduria.
Parkinsonism & related disordersThe outcome of 41 Late-Diagnosed Turkish GA-1 Patients: A Candidate for the Turkish NBS.
NeuropediatricsMRI of 3-hydroxyisobutyryl-CoA hydrolase (HIBCH) deficiency.
Radiology case reportsEnteral tube feeding in patients receiving dietary treatment for metabolic diseases: A retrospective analysis in a large French cohort.
Molecular genetics and metabolism reportsTwo cases of carbonic anhydrase VA deficiency-An ultrarare metabolic decompensation syndrome presenting with hyperammonemia, lactic acidosis, ketonuria, and good clinical outcome.
JIMD reportsHigh frequency of biotinidase deficiency in Italian population identified by newborn screening.
Molecular genetics and metabolism reportsPrecocious puberty in a girl with 3-methylglutaconic aciduria type 1 (3-MGA-I) due to a novel AUH gene mutation.
Molecular genetics and metabolism reportstrans-3-Methylglutaconyl CoA isomerization-dependent protein acylation.
Biochemical and biophysical research communicationsInconsistencies in the Nutrition Management of Glutaric Aciduria Type 1: An International Survey.
NutrientsPhenotypic spectrum of short-chain enoyl-Coa hydratase-1 (ECHS1) deficiency.
European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology SocietyIntrathecal Baclofen for Hypertonia Secondary to Glutaric Aciduria Type I.
CureusOrganic acidurias: Major gaps, new challenges, and a yet unfulfilled promise.
Journal of inherited metabolic diseaseImpaired lipolysis in propionic acidemia: A new metabolic myopathy?
JIMD reports2-methylacetoacetyl-coenzyme A thiolase (beta-ketothiolase) deficiency: one disease - two pathways.
Orphanet journal of rare diseasesCardiac phenotype in propionic acidemia - Results of an observational monocentric study.
Molecular genetics and metabolism3-hydroxy-3-methylglutaryl-coenzyme A lyase deficiency: one disease - many faces.
Orphanet journal of rare diseasesDiagnosis of inherited metabolic disorders by selective metabolite testing: three years' experience at a tertiary care center in Rawalpindi.
JPMA. The Journal of the Pakistan Medical AssociationA 7-year-old Boy with Hand Tremors and a Novel Mutation for L-2-hydroxyglutaric Aciduria.
Balkan journal of medical genetics : BJMGd-Glycerate kinase deficiency in a neuropediatric patient.
Brain & developmentUntargeted Metabolomics-Based Screening Method for Inborn Errors of Metabolism using Semi-Automatic Sample Preparation with an UHPLC- Orbitrap-MS Platform.
Metabolites3-Methylglutaric acid in energy metabolism.
Clinica chimica acta; international journal of clinical chemistryInborn Errors of Metabolism in the Era of Untargeted Metabolomics and Lipidomics.
MetabolitesClinicoradiological Spectrum of L-2-Hydroxy Glutaric Aciduria: Typical and Atypical Findings in an Indian Cohort.
Journal of clinical imaging scienceAcylcarnitine profiling by low-resolution LC-MS.
PloS oneThe Therapeutic Hypothermia in Treatment of Hyperammonemic Encephalopathy due to Urea Cycle Disorders and Organic Acidemias.
Klinische Padiatrie3-Hydroxyisobutyryl-CoA hydrolase deficiency in an Iranian child with novel HIBCH compound heterozygous mutations.
Clinical case reportsDifficulties in the dietary management of a girl with two diseases requiring a special diet.
Developmental period medicineGlutaric Aciduria Type 1 with Microcephaly: Masquerading as Spastic Cerebral Palsy.
Journal of pediatric neurosciencesUnique neuroradiological findings in propionic acidemia.
Radiology case reportsTwo novel L2HGDH mutations identified in a rare Chinese family with L-2-hydroxyglutaric aciduria.
BMC medical geneticsInborn errors of metabolism associated with hyperglycaemic ketoacidosis and diabetes mellitus: narrative review.
Sudanese journal of paediatricsEffect of carglumic acid with or without ammonia scavengers on hyperammonaemia in acute decompensation episodes of organic acidurias.
Orphanet journal of rare diseasesBiochemical and molecular characterization of 3-Methylcrotonylglycinuria in an Italian asymptomatic girl.
Genetics and molecular biologyLong-term liver disease in methylmalonic and propionic acidemias.
Molecular genetics and metabolism[Screening for newborn organic aciduria in Zhejiang province:prevalence, outcome and follow-up].
Zhejiang da xue xue bao. Yi xue ban = Journal of Zhejiang University. Medical sciencesGenetic Screening of Selected Disease-Causing Mutations in Glutaryl-CoA Dehydrogenase Gene among Indian Patients with Glutaric Aciduria Type I.
Journal of pediatric geneticsGlutaric Aciduria Type 1 and Acute Renal Failure: Case Report and Suggested Pathomechanisms.
JIMD reports3-Hydroxy-3-methylglutaryl-coenzyme A lyase deficiency: Clinical presentation and outcome in a series of 37 patients.
Molecular genetics and metabolismElevated glutaric acid levels in Dhtkd1-/Gcdh- double knockout mice challenge our current understanding of lysine metabolism.
Biochimica et biophysica acta. Molecular basis of diseaseBarth Syndrome: Connecting Cardiolipin to Cardiomyopathy.
LipidsBarth syndrome: A life-threatening disorder caused by abnormal cardiolipin remodeling.
Journal of rare diseases research & treatment"Classical organic acidurias": diagnosis and pathogenesis.
Clinical and experimental medicine3-Methylcrotonyl-CoA carboxylase deficiency: Mutational spectrum derived from comprehensive newborn screening.
GeneThe M405V allele of the glutaryl-CoA dehydrogenase gene is an important marker for glutaric aciduria type I (GA-I) low excretors.
Molecular genetics and metabolismSevere Neonatal Presentation of Mitochondrial Citrate Carrier (SLC25A1) Deficiency.
JIMD reportsOn the origin of 3-methylglutaconic acid in disorders of mitochondrial energy metabolism.
Journal of inherited metabolic diseaseAngelman syndrome and isovaleric acidemia: What is the link?
Molecular genetics and metabolism reportsPilot Experience with an External Quality Assurance Scheme for Acylcarnitines in Plasma/Serum.
JIMD reportsExpanding the phenotype in aminoacylase 1 (ACY1) deficiency: characterization of the molecular defect in a 63-year-old woman with generalized dystonia.
Metabolic brain diseasePrimary and maternal 3-methylcrotonyl-CoA carboxylase deficiency: insights from the Israel newborn screening program.
Journal of inherited metabolic diseaseValproate therapy exacerbating intermediate phenotype of methylmalonic aciduria.
Journal of pediatric neurosciencesCardiolipin fingerprinting of leukocytes by MALDI-TOF/MS as a screening tool for Barth syndrome.
Journal of lipid researchNeurometabolic Disorders-Related Early Childhood Epilepsy: A Single-Center Experience in Saudi Arabia.
Pediatrics and neonatologyClinical manifestations and enzymatic activities of mitochondrial respiratory chain complexes in Pearson marrow-pancreas syndrome with 3-methylglutaconic aciduria: a case report and literature review.
European journal of pediatricsFive novel SUCLG1 mutations in three Chinese patients with succinate-CoA ligase deficiency noticed by mild methylmalonic aciduria.
Brain & developmentEffect of a 5-mo nutritional intervention on nutritional status and quality of life for patient with 3-hydroxyisobutyryl-coenzyme A hydrolase deficiency: A case report.
Nutrition (Burbank, Los Angeles County, Calif.)Anesthesia and organic aciduria: is the use of lactated Ringer's solution absolutely contraindicated?
Paediatric anaesthesiaBiochemical abnormalities in Pearson syndrome.
American journal of medical genetics. Part AAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Acidúria orgânica.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Acidúria orgânica
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Brain morphometry and cognition in late-onset glutaric aciduria type 1: scoping review and novel insights from a case report.Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology· 2026· PMID 41779049mais citado
- Nutritional Management in Severe Methylmalonic and Propionic Acidemias: How Much Medical Food Is Too Much?
- Global Longitudinal Strain Alteration of the Left Ventricle in Children with Organic Aciduria: Cardiac Disease in Organic Aciduria.
- The Association Between Metabolomic and Usual Biochemical Data Helps to Detect Insulin Resistance.
- Clinical and neuroradiologic spectrum of glutaric acidemia type 1 in children: insights from a retrospective cohort in Guangdong Province, China.
- Whole exome sequencing in pediatric hyperammonemia: significant diagnostic yield and identification of three novel variants.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:289899(Orphanet)
- MONDO:0000688(MONDO)
- GARD:9433(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q1547640(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
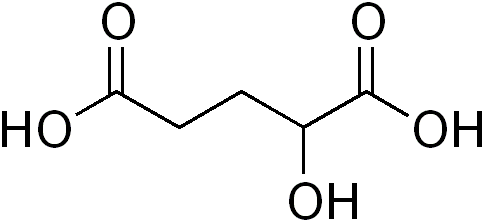
Acidúria orgânica
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos (literatura)
- fonte: Orphanet