A displasia mandibuloacral (MAD) é uma doença óssea genética rara caracterizada por atraso no crescimento, desenvolvimento pós-natal de anomalias craniofaciais, incluindo hipoplasia mandibular, osteólise acral progressiva, pigmentação manchada ou irregular, atrofia da pele e lipodistrofia parcial ou generalizada.
Introdução
O que você precisa saber de cara
A displasia mandibuloacral (MAD) é uma doença óssea genética rara caracterizada por atraso no crescimento, desenvolvimento pós-natal de anomalias craniofaciais, incluindo hipoplasia mandibular, osteólise acral progressiva, pigmentação manchada ou irregular, atrofia da pele e lipodistrofia parcial ou generalizada.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 30 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 97 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Transmembrane metalloprotease whose catalytic activity is critical for processing lamin A/LMNA on the inner nuclear membrane and clearing clogged translocons on the endoplasmic reticulum (PubMed:33293369, PubMed:33315887). Proteolytically removes the C-terminal three residues of farnesylated proteins (PubMed:33293369, PubMed:33315887). Also plays an antiviral role independently of its protease activity by restricting enveloped RNA and DNA viruses, including influenza A, Zika, Ebola, Sindbis, ves
Endoplasmic reticulum membraneNucleus inner membraneEarly endosome membraneLate endosome membrane
Mandibuloacral dysplasia with type B lipodystrophy
A form of mandibuloacral dysplasia, a rare progeroid disorder with clinical and genetic heterogeneity, characterized by growth retardation, craniofacial dysmorphic features due to distal bone resorption, musculoskeletal and skin abnormalities associated with lipodystrophy. MADB is a disease characterized by mandibular and clavicular hypoplasia, acroosteolysis, delayed closure of the cranial suture, joint contractures, and generalized lipodystrophy with loss of subcutaneous fat from the extremities, face, neck and trunk.
Lamins are intermediate filament proteins that assemble into a filamentous meshwork, and which constitute the major components of the nuclear lamina, a fibrous layer on the nucleoplasmic side of the inner nuclear membrane (PubMed:10080180, PubMed:10580070, PubMed:10587585, PubMed:10814726, PubMed:11799477, PubMed:12075506, PubMed:12927431, PubMed:15317753, PubMed:18551513, PubMed:18611980, PubMed:2188730, PubMed:22431096, PubMed:2344612, PubMed:23666920, PubMed:24741066, PubMed:31434876, PubMed:
Nucleus laminaNucleus envelopeNucleus, nucleoplasmNucleus matrixNucleus speckle
Emery-Dreifuss muscular dystrophy 2, autosomal dominant
A form of Emery-Dreifuss muscular dystrophy, a degenerative myopathy characterized by weakness and atrophy of muscle without involvement of the nervous system, early contractures of the elbows, Achilles tendons and spine, and cardiomyopathy associated with cardiac conduction defects.
Variantes genéticas (ClinVar)
967 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 304 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
8 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Displasia mandíbulo-acral
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
A Case of Restrictive Dermopathy With Atypical Cardiac Anomalies and a Novel ZMPSTE24 Variant.
Restrictive Dermopathy (RD, OMIM #275210) is an ultra-rare, lethal genodermatosis caused by defects in lamins and related proteins. RD is caused by biallelic pathogenic variants in ZMPSTE24, which encodes zinc metallopeptidase ZMPSTE24, an enzyme essential for processing prelamin A, the precursor of lamin A. While null variants leading to a total loss of prelamin A processing have been related to neonatally lethal RD, variants preserving residual prelamin A processing function cause an allelic disorder called Mandibuloacral Dysplasia Type B (MAD-B). RD is characterized by taut translucent skin, visible superficial vessels, joint contractures, and dysmorphic features, with death usually occurring within the first month of life. Cardiac anomalies, including ASD and PDA, have been reported in a few patients, most of whom were not genetically confirmed, and transposition of the great arteries (TGA) has been described only once, also without molecular confirmation. We report a patient with restrictive dermatopathy (RD) presenting with double outlet right ventricle (DORV) and pulmonary valve atresia which have not been previously reported in association with RD. Exome Sequencing (ES) was performed, revealing a novel homozygous splice-site variant in ZMPSTE24 (c.1203 + 1G>T). Segregation analysis was performed in the mother and two siblings. To our knowledge, DORV has not previously been reported in an RD patient. This case expands the genotypic spectrum of RD and suggests a possible link with complex cardiac malformations.
Founder Pathogenic Variant in LMNA with Diverse Phenotypic Manifestations in Mandibuloacral Dysplasia: Insights from a Turkish Cohort.
Mandibuloacral dysplasia (MAD) is a rare genetic disorder characterized by distinctive skeletal abnormalities, metabolic issues, and skin changes, often linked to pathogenic variants in the LMNA gene, which encodes lamin A/C. This study investigates a specific founder mutation within a Turkish cohort and explores its impact on phenotypic expressivity. We conducted a comprehensive analysis involving genetic testing for LMNA variants in patients diagnosed with MAD. Clinical evaluations documented a wide range of phenotypic features, including facial dysmorphism, skeletal anomalies, and metabolic abnormalities. We also collected family histories to assess inheritance patterns and potential environmental influences. Our findings identified a common founder mutation in the LMNA gene among the cohort, which was present in a significant percentage of participants. Notably, phenotypic expressivity varied significantly, with some individuals exhibiting classic MAD features, while others showed atypical manifestations, such as additional endocrine disorders and variable severity of skeletal anomalies. This variability underscores the complexity of the genotype-phenotype relationship. This study highlights the significance of the founder mutation in LMNA and its diverse phenotypic outcomes in MAD. Our results contribute to the understanding of how genetic mutations can lead to a spectrum of clinical presentations, emphasizing the necessity for personalized clinical approaches in managing this condition. Further research is warranted to elucidate the underlying mechanisms of phenotypic variability and to improve diagnostic and therapeutic strategies.
MAM-STAT3-Driven Mitochondrial Ca+2 Upregulation Contributes to Immunosenescence in Type A Mandibuloacral Dysplasia Patients.
Individuals with homozygous laminA/C p.R527C mutations manifest a severe form of Mandibuloacral dysplasia-(MAD) and exhibit overlapping progeroid symptoms, for which the underlying molecular pathology remains unknown. Herein, it is shown that MAD patients achieved inflammaging with different pro-inflammatory cytokines compared to progeria-(HGPS) patient. Characterization of MAD iPSC-derived Mesenchymal stem cells (MAD-iMSC) uncovers deregulated mitochondrial Ca+2 as the primary cause of inflammaging, mediated through inflammasome formation rather than the cGAS-STING pathway. Moreover, MAD-iMSCs extracellular vesicles (EVs) can also upregulate mitochondrial Ca+2 in healthy cells. This deregulated Ca+2 homeostasis is indirectly mediated by mitochondrial calcium mediator, signal transducer, and activator of transcription-3 (STAT3), situated on the mitochondrial associated membrane (MAM). Inflammaging is mitigated by various FDA-approved MAM-STAT3 upstream inhibitors, such as (Tocilizumab) or by correcting R527C mutation with CRISPR/CAS9. These results provide new insights into MAD disease and propose targeting defective mitochondrial Ca+2 homeostasis as a promising therapy for reversing immunosenescence.
P18 ZMPSTE24 variant with the lethal phenotype of restrictive dermopathy.
Restrictive dermopathy (RD) is a rare, lethal, congenital autosomal recessive laminopathy syndrome characterized by generalised tight translucent skin, dysmorphic facies, arthrogryposis multiplex congenita, and pulmonary hypoplasia. It is caused by mutations in either ZMPSTE24 or LMNA. Variants in these genes, which disrupt the production of lamin A, and hence the structural integrity of the nuclear envelope, can also cause survivable syndromes such as mandibuloacral dysplasia (MAD). We report the first son of non-related Caucasian parents delivered via emergency LSCS at 34 weeks following preeclampsia, gestational diabetes and polyhydramnios. Extraction was difficult due to arthrogryposis. At birth (weight 1.65 kg), he showed the typical features of RD. Genetic testing revealed a homozygous pathogenic ZMPSTE24 frameshift variant c.1085dup p. (Leu362PhefsTer19) previously reported in mandibuloacral dysplasia (Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet 2003;12:1995-2001). Parents were heterozygous carriers. The baby received palliative care and died on day 2. The longest survival in RD is 120 days, in contrast to other laminopathies. Similar or identical mutation within the LMNA gene produces varied phenotypes which expands the spectrum. Currently, there is no curative therapy for RD. Our main aim to diagnose this condition is to offer counselling, genetic testing for future pregnancies, prevent unnecessary interventions and support the family through compassion and dignity.
Severe cardiac valvular calcification in two Chinese brothers with mandibuloacral dysplasia type A: a case report.
Mandibuloacral dysplasia type A (MADA) is a rare progeroid syndrome associated with mutations in the Lamin A/C (LMNA) gene, primarily affecting skeletal, cutaneous, and adipose tissues. While certain LMNA gene mutations are known to cause cardiomyopathy and conduction system disease, severe early-onset calcific valvular heart disease is not conventionally considered a typical feature of MADA. This report describes two brothers from a consanguineous Han Chinese family who presented with classical MADA phenotypes alongside severe, early-onset cardiac valvular calcification. Genetic investigation revealed that both affected brothers carried a homozygous missense mutation, c.785A > G (p.Glu262Gly), in the LMNA gene. The elder brother, aged 41, successfully underwent transcatheter aortic valve implantation (TAVI) due to severe aortic valve stenosis. This finding represents the first association, to our knowledge, between the homozygous LMNA c.785A > G (p.Glu262Gly) mutation and significant severe early-onset cardiac valvular calcification manifesting as a prominent feature within the MADA phenotype, thus expanding the clinical spectrum associated with MADA and this specific LMNA variant. This case highlights a potentially underrecognized cardiovascular manifestation in MADA patients and underscores the importance of comprehensive cardiac assessment in affected individuals, particularly those from consanguineous families.
Publicações recentes
A Case of Restrictive Dermopathy With Atypical Cardiac Anomalies and a Novel ZMPSTE24 Variant.
💬 OpiniãoP18 ZMPSTE24 variant with the lethal phenotype of restrictive dermopathy.
Severe cardiac valvular calcification in two Chinese brothers with mandibuloacral dysplasia type A: a case report.
Mtx2 requirement for craniofacial morphogenesis with implications for mandibuloacral dysplasia.
Homozygous LMNA c.1579C>T mutation manifesting as mandibuloacral dysplasia type A in a pediatric patient.
📚 EuropePMC81 artigos no totalmostrando 61
A Case of Restrictive Dermopathy With Atypical Cardiac Anomalies and a Novel ZMPSTE24 Variant.
American journal of medical genetics. Part AP18 ZMPSTE24 variant with the lethal phenotype of restrictive dermopathy.
The British journal of dermatologySevere cardiac valvular calcification in two Chinese brothers with mandibuloacral dysplasia type A: a case report.
Frontiers in cardiovascular medicineMtx2 requirement for craniofacial morphogenesis with implications for mandibuloacral dysplasia.
Biochemical and biophysical research communicationsHomozygous LMNA c.1579C>T mutation manifesting as mandibuloacral dysplasia type A in a pediatric patient.
Indian journal of dermatology, venereology and leprologyFounder Pathogenic Variant in LMNA with Diverse Phenotypic Manifestations in Mandibuloacral Dysplasia: Insights from a Turkish Cohort.
Journal of clinical research in pediatric endocrinologyA Case of Mandibuloacral Dysplasia Progeroid Syndrome Presented as In Utero Growth Restriction.
Journal of medical ultrasoundRare ZMPSTE24 variants increase risk of hypertriglyceridemia and metabolic syndrome.
European journal of endocrinologyMetaxin-2 tunes mitochondrial transportation and neuronal function in Drosophila.
GeneticsMAM-STAT3-Driven Mitochondrial Ca+2 Upregulation Contributes to Immunosenescence in Type A Mandibuloacral Dysplasia Patients.
Advanced science (Weinheim, Baden-Wurttemberg, Germany)Disorganized chromatin hierarchy and stem cell aging in a male patient of atypical laminopathy-based progeria mandibuloacral dysplasia type A.
Nature communicationsValidation of metaxin-2 deficient C. elegans as a model for MandibuloAcral Dysplasia associated to mtx-2 (MADaM) syndrome.
Communications biologyRadiological insights into mandibuloacral dysplasia: A case report.
Radiology case reportsAcrofacial dysmorphism and lipodystrophy: unveiling mandibuloacral dysplasia in a case of young onset diabetes mellitus.
BMJ case reportsLipodystrophic Laminopathies: From Dunnigan Disease to Progeroid Syndromes.
International journal of molecular sciencesNavigating Lipodystrophy: Insights from Laminopathies and Beyond.
International journal of molecular sciencesA rare LMNA missense mutation causing a severe phenotype of mandibuloacral dysplasia type A: a case report.
Revista paulista de pediatria : orgao oficial da Sociedade de Pediatria de Sao PauloMandibuloacral dysplasia type A.
BMJ case reportsCase report: A novel splice-site mutation of MTX2 gene caused mandibuloacral dysplasia progeroid syndrome: the first report from China and literature review.
Frontiers in endocrinologyThe farnesyl transferase inhibitor (FTI) lonafarnib improves nuclear morphology in ZMPSTE24-deficient fibroblasts from patients with the progeroid disorder MAD-B.
Nucleus (Austin, Tex.)Prelamin A and ZMPSTE24 in premature and physiological aging.
Nucleus (Austin, Tex.)Multi-omic analysis of mandibuloacral dysplasia type A patient iPSC-derived MSC senescence reveals miR-311 as a novel biomarker for MSC senescence.
Human molecular geneticsImpact of Combined Baricitinib and FTI Treatment on Adipogenesis in Hutchinson-Gilford Progeria Syndrome and Other Lipodystrophic Laminopathies.
CellsAnalysis of a non-lethal biallelic frameshift mutation in ZMPSTE24 reveals utilization of alternative translation initiation codons.
Clinical geneticsDiagnosis and genetic analysis of a case with mandibuloacral dysplasia type B due to compound heterozygous mutations of the ZMPSTE24 gene.
Yi chuan = HereditasHutchinson-Gilford Syndrome (Progeria) with Heterozygous Mutation in the LMNA Gene-ENST00000368300.9 Presenting with Mandibuloacral Dysplasia and Acrogeroid Features-Overlap of Premature Aging Syndromes.
Indian dermatology online journalA novel MTX2 gene splice site variant resulting in exon skipping, causing the recently described mandibuloacral dysplasia progeroid syndrome.
American journal of medical genetics. Part AAn exceptional biallelic N-terminal frame shift mutation in ZMPSTE24 leads to non-lethal progeria due to possible utilization of a downstream alternative start codon.
GeneTwo Decades after Mandibuloacral Dysplasia Discovery: Additional Cases and Comprehensive View of Disease Characteristics.
GenesMandibuloacral dysplasia in a young Vietnamese girl caused by homozygous missense variant c.1579C>T in the LMNA gene with progeria and severe skin lesions.
JAAD case reportsTranscatheter Aortic Valve Replacement in a Young Patient With Mandibuloacral Dysplasia.
JACC. Case reportsMutations Involved in Premature-Ageing Syndromes.
The application of clinical geneticsAtypical progeroid syndrome (p.E262K LMNA mutation): a rare cause of short stature and osteoporosis.
Endocrinology, diabetes & metabolism case reportsFunctional analysis of POLD1 p.ser605del variant: the aging phenotype of MDPL syndrome is associated with an impaired DNA repair capacity.
AgingCutaneous and metabolic defects associated with nuclear abnormalities in a transgenic mouse model expressing R527H lamin A mutation causing mandibuloacral dysplasia type A (MADA) syndrome.
Acta myologica : myopathies and cardiomyopathies : official journal of the Mediterranean Society of MyologyMandibuloacral dysplasia type A in five tunisian patients.
European journal of medical geneticsAuthor Correction: Loss of MTX2 causes mandibuloacral dysplasia and links mitochondrial dysfunction to altered nuclear morphology.
Nature communicationsNovel clinical features and pleiotropic effect in three unrelated patients with LMNA variant.
Clinical dysmorphologyMultisystem Progeroid Syndrome With Lipodystrophy, Cardiomyopathy, and Nephropathy Due to an LMNA p.R349W Variant.
Journal of the Endocrine SocietyLoss of MTX2 causes mandibuloacral dysplasia and links mitochondrial dysfunction to altered nuclear morphology.
Nature communicationsLamin A involvement in ageing processes.
Ageing research reviewsA novel autosomal recessive lipodystrophy syndrome due to homozygous LMNA variant.
Journal of medical geneticsMandibuloacral dysplasia type B (MADB): a cohort of eight patients from Suriname with a homozygous founder mutation in ZMPSTE24 (FACE1), clinical diagnostic criteria and management guidelines.
Orphanet journal of rare diseasesMandibuloacral dysplasia with type B lipodystrophy in a patient from Chile.
American journal of medical genetics. Part ATissue-Specific Influence of Lamin A Mutations on Notch Signaling and Osteogenic Phenotype of Primary Human Mesenchymal Cells.
CellsThe Cutting Edge: The Role of mTOR Signaling in Laminopathies.
International journal of molecular sciencesProteomic Evidence of Biological Aging in a Child with a Compound Heterozygous ZMPSTE24 Mutation.
Proteomics. Clinical applicationsLamins and bone disorders: current understanding and perspectives.
OncotargetZMPSTE24 missense mutations that cause progeroid diseases decrease prelamin A cleavage activity and/or protein stability.
Disease models & mechanismsPhenotypic heterogeneity of ZMPSTE24 deficiency.
American journal of medical genetics. Part AA Novel Generalized Lipodystrophy-Associated Progeroid Syndrome Due to Recurrent Heterozygous LMNA p.T10I Mutation.
The Journal of clinical endocrinology and metabolismMandibuloacral dysplasia: A premature ageing disease with aspects of physiological ageing.
Ageing research reviewsProgeroid syndrome patients with ZMPSTE24 deficiency could benefit when treated with rapamycin and dimethylsulfoxide.
Cold Spring Harbor molecular case studiesAntisense-Based Progerin Downregulation in HGPS-Like Patients' Cells.
CellsFailure of ossification of the occipital bone in mandibuloacral dysplasia type B.
American journal of medical genetics. Part ANovel LMNA mutations cause an aggressive atypical neonatal progeria without progerin accumulation.
Journal of medical geneticsMandibuloacral dysplasia and LMNA A529V mutation in Turkish patients with severe skeletal changes and absent breast development.
Clinical dysmorphologyA mutation abolishing the ZMPSTE24 cleavage site in prelamin A causes a progeroid disorder.
Journal of cell scienceA novel homozygous LMNA mutation (p.Met540Ile) causes mandibuloacral dysplasia type A.
GeneModulation of TGFbeta 2 levels by lamin A in U2-OS osteoblast-like cells: understanding the osteolytic process triggered by altered lamins.
OncotargetMandibuloacral dysplasia type B in an infant: a rare progeroid genodermatosis.
JAMA dermatologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Displasia mandíbulo-acral.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Displasia mandíbulo-acral
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- A Case of Restrictive Dermopathy With Atypical Cardiac Anomalies and a Novel ZMPSTE24 Variant.
- Founder Pathogenic Variant in LMNA with Diverse Phenotypic Manifestations in Mandibuloacral Dysplasia: Insights from a Turkish Cohort.
- MAM-STAT3-Driven Mitochondrial Ca+2 Upregulation Contributes to Immunosenescence in Type A Mandibuloacral Dysplasia Patients.
- P18 ZMPSTE24 variant with the lethal phenotype of restrictive dermopathy.
- Severe cardiac valvular calcification in two Chinese brothers with mandibuloacral dysplasia type A: a case report.
- Mtx2 requirement for craniofacial morphogenesis with implications for mandibuloacral dysplasia.
- Homozygous LMNA c.1579C>T mutation manifesting as mandibuloacral dysplasia type A in a pediatric patient.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2457(Orphanet)
- MONDO:0016584(MONDO)
- GARD:11893(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q16968886(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
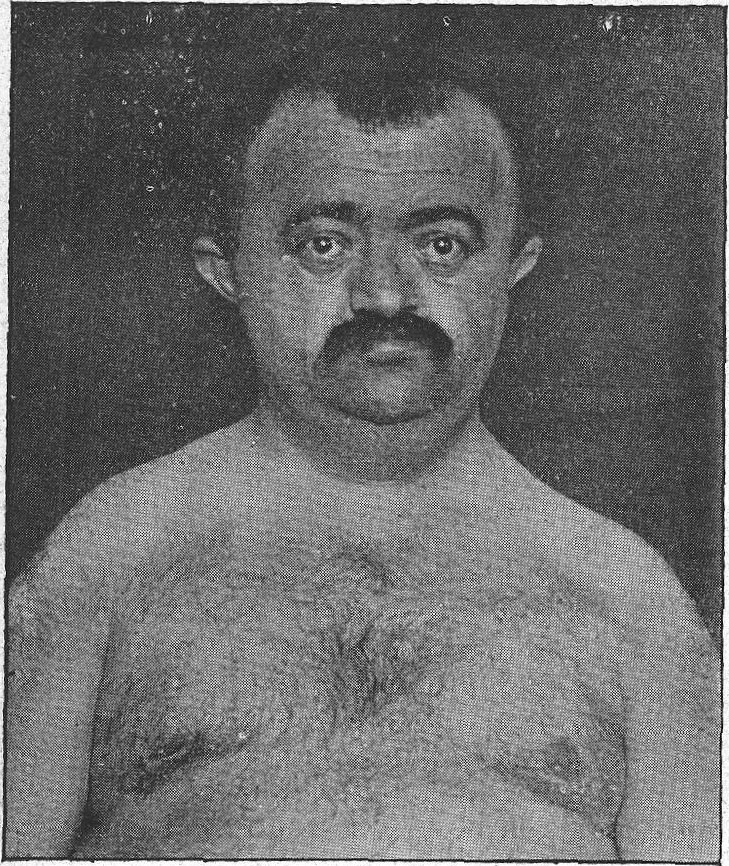
Displasia mandíbulo-acral
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata