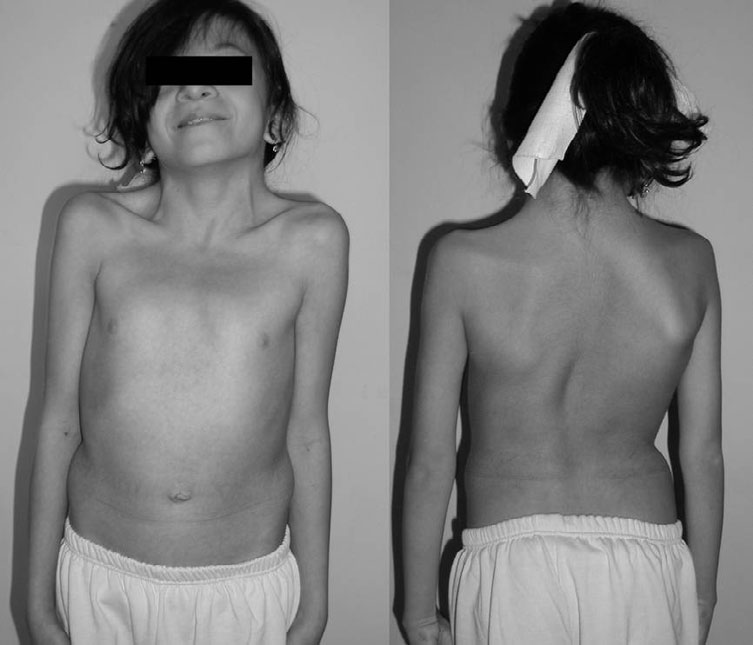
A síndrome relacionada ao CBL é uma condição genética causada por alterações genéticas prejudiciais no gene CBL. Devido ao mecanismo proposto que indica a relação do gene CBL com a via de sinalização celular RAS-MAPK e à apresentação da doença ser parecida com a das RASopatias, a síndrome relacionada ao CBL deve ser considerada uma RASopatia. Embora haja uma grande variedade de características, em geral, pacientes com a síndrome relacionada ao CBL podem apresentar atraso no desenvolvimento, deficiência intelectual, alterações no desenvolvimento neurológico, problemas linfáticos que surgem antes do nascimento, malformações cardíacas e problemas nos vasos sanguíneos, especialmente no cérebro (como a doença de Moya-Moya). Também podem ter características faciais e cranianas típicas de RASopatias, tônus muscular baixo (hipotonia), dificuldades para se alimentar, inchaço nas pernas, alterações nos músculos, ossos e sistema respiratório do tórax, características da pele como manchas café com leite, problemas imunológicos e no sangue, e uma maior propensão a certos tipos de tumores, como a leucemia mielomonocítica juvenil (LMJM), que geralmente regride sozinha. Vale ressaltar que o risco de outros tipos de tumores além da LMJM ainda não foi completamente avaliado. Devido à ampla variedade de como essas e outras características se manifestam em pacientes com alterações no gene CBL, os especialistas atualmente definem essas condições fazendo referência ao gene causador, o CBL.
Introdução
O que você precisa saber de cara
A síndrome relacionada ao CBL é uma condição genética causada por alterações genéticas prejudiciais no gene CBL. Devido ao mecanismo proposto que indica a relação do gene CBL com a via de sinalização celular RAS-MAPK e à apresentação da doença ser parecida com a das RASopatias, a síndrome relacionada ao CBL deve ser considerada uma RASopatia. Embora haja uma grande variedade de características, em geral, pacientes com a síndrome relacionada ao CBL podem apresentar atraso no desenvolvimento, deficiência intelectual, alterações no desenvolvimento neurológico, problemas linfáticos que surgem antes do nascimento, malformações cardíacas e problemas nos vasos sanguíneos, especialmente no cérebro (como a doença de Moya-Moya). Também podem ter características faciais e cranianas típicas de RASopatias, tônus muscular baixo (hipotonia), dificuldades para se alimentar, inchaço nas pernas, alterações nos músculos, ossos e sistema respiratório do tórax, características da pele como manchas café com leite, problemas imunológicos e no sangue, e uma maior propensão a certos tipos de tumores, como a leucemia mielomonocítica juvenil (LMJM), que geralmente regride sozinha. Vale ressaltar que o risco de outros tipos de tumores além da LMJM ainda não foi completamente avaliado. Devido à ampla variedade de como essas e outras características se manifestam em pacientes com alterações no gene CBL, os especialistas atualmente definem essas condições fazendo referência ao gene causador, o CBL.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 19 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 50 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição.
Curadoria gene-doença
fontes oficiaisE3 ubiquitin-protein ligase that acts as a negative regulator of many signaling pathways by mediating ubiquitination of cell surface receptors (PubMed:10514377, PubMed:11896602, PubMed:14661060, PubMed:14739300, PubMed:15190072, PubMed:17509076, PubMed:18374639, PubMed:19689429, PubMed:21596750, PubMed:28381567, PubMed:40101708). Accepts ubiquitin from specific E2 ubiquitin-conjugating enzymes, and then transfers it to substrates promoting their degradation by the proteasome (PubMed:10514377, Pu
CytoplasmCell membraneCell projection, ciliumGolgi apparatus
Noonan syndrome-like disorder with or without juvenile myelomonocytic leukemia
A syndrome characterized by a phenotype reminiscent of Noonan syndrome. Clinical features are highly variable, including facial dysmorphism, short neck, developmental delay, hyperextensible joints and thorax abnormalities with widely spaced nipples. The facial features consist of triangular face with hypertelorism, large low-set ears, ptosis, and flat nasal bridge. Some patients manifest cardiac defects. Some have an increased risk for certain malignancies, particularly juvenile myelomonocytic leukemia.
Variantes genéticas (ClinVar)
282 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 6 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
20 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Doença semelhante à síndrome de Noonan com leucemia mielomonocítica juvenil
Centros de Referência SUS
24 centros habilitados pelo SUS para Doença semelhante à síndrome de Noonan com leucemia mielomonocítica juvenil
Centros para Doença semelhante à síndrome de Noonan com leucemia mielomonocítica juvenil
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
Somatic variation as an incidental finding in the pediatric next-generation sequencing era.
The methodologic approach used in next-generation sequencing (NGS) affords a high depth of coverage in genomic analysis. Inherent in the nature of genomic testing, there exists potential for identifying genomic findings that are incidental or secondary to the indication for clinical testing, with the frequency dependent on the breadth of analysis and the tissue sample under study. The interpretation and management of clinically meaningful incidental genomic findings is a pressing issue particularly in the pediatric population. Our study describes a 16-mo-old male who presented with profound global delays, brain abnormality, progressive microcephaly, and growth deficiency, as well as metopic craniosynostosis. Clinical exome sequencing (ES) trio analysis revealed the presence of two variants in the proband. The first was a de novo variant in the PPP2R1A gene (c.773G > A, p.Arg258His), which is associated with autosomal dominant (AD) intellectual disability, accounting for the proband's clinical phenotype. The second was a recurrent hotspot variant in the CBL gene (c.1111T > C, p.Tyr371His), which was present at a variant allele fraction of 11%, consistent with somatic variation in the peripheral blood sample. Germline pathogenic variants in CBL are associated with AD Noonan syndrome-like disorder with or without juvenile myelomonocytic leukemia. Molecular analyses using a different tissue source, buccal epithelial cells, suggest that the CBL alteration may represent a clonal population of cells restricted to leukocytes. This report highlights the laboratory methodologic and interpretative processes and clinical considerations in the setting of acquired variation detected during clinical ES in a pediatric patient.
A case of splenomegaly in CBL syndrome.
We present a child with unexplained splenomegaly to highlight this feature as a presenting sign of the RASopathy CBL syndrome and to draw attention to the power and utility of next generation genomic sequencing for providing rapid diagnosis and critical information to guide care in the pediatric clinical setting. A 7-year-old boy presented with unexplained splenomegaly, attention deficit hyperactivity disorder, mild learning difficulties, easy bruising, mild thrombocytopenia, and subtle dysmorphic features. Extensive haematological testing including a bone marrow biopsy showed mild megaloblastoid erythropoiesis and borderline fibrosis. There were no haematological cytogenetic anomalies or other haematological pathology to explain the splenomegaly. Metabolic testing and chromosomal microarray were unremarkable. Trio whole-exome sequencing (WES) identified a pathogenic de novo heterozygous germline CBL variant (c.1111T > C, p.Y371H), previously reported to cause CBL syndrome and implicated in development of juvenile myelomonocytic leukemia (JMML). CBL syndrome (more formally known as "Noonan-syndrome-like disorder with or without juvenile myelomonocytic leukemia") has overlapping features to Noonan syndrome with significant variability. CBL syndrome and other RASopathy disorders-including Noonan syndrome, neurofibromatosis 1, and Costello syndrome-are important to recognize as these are associated with a cancer-predisposition. CBL syndrome carries a very high risk for JMML, thus accurate diagnosis is of utmost importance. The diagnosis of CBL syndrome in this patient would not have been possible based on clinical features alone. Through WES, a specific genetic diagnosis was made, allowing for an optimized management and surveillance plan, illustrating the power of genomics in clinical practice.
Adults with germline CBL mutation complicated with juvenile myelomonocytic leukemia at infancy.
Juvenile myelomonocytic leukemia (JMML) appears to be a life-threatening disease and showed poor prognosis even after hematopoietic stem cell transplantation (HSCT) because of high relapse rate. On the other hand, recent molecular analysis revealed the heterogeneity of JMML. Here we report that two JMML patients survived >20 years without HSCT and both patients had uniparental disomy of 11q23 where CBL is located without the phenomenon found in neither Noonan syndrome nor Noonan syndrome-like disorder. We think that some JMML patients with CBL mutation might show the good prognosis in later life after remission of JMML.
Publicações recentes
Somatic variation as an incidental finding in the pediatric next-generation sequencing era.
🥉 Relato de casoA case of splenomegaly in CBL syndrome.
Adults with germline CBL mutation complicated with juvenile myelomonocytic leukemia at infancy.
Hydrops, fetal pleural effusions and chylothorax in three patients with CBL mutations.
📚 EuropePMCmostrando 3
Somatic variation as an incidental finding in the pediatric next-generation sequencing era.
Cold Spring Harbor molecular case studiesA case of splenomegaly in CBL syndrome.
European journal of medical geneticsAdults with germline CBL mutation complicated with juvenile myelomonocytic leukemia at infancy.
Journal of human geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Doença semelhante à síndrome de Noonan com leucemia mielomonocítica juvenil.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Doença semelhante à síndrome de Noonan com leucemia mielomonocítica juvenil
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Somatic variation as an incidental finding in the pediatric next-generation sequencing era.
- A case of splenomegaly in CBL syndrome.
- Adults with germline CBL mutation complicated with juvenile myelomonocytic leukemia at infancy.
- Noonan Syndrome.
- Hydrops, fetal pleural effusions and chylothorax in three patients with CBL mutations.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:363972(Orphanet)
- OMIM OMIM:613563(OMIM)
- MONDO:0013308(MONDO)
- GARD:17577(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55784010(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Doença semelhante à síndrome de Noonan com leucemia mielomonocítica juvenil
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata