A epidermólise bolhosa distrófica recessiva grave e generalizada (RDEB-sev gen) é o subtipo mais grave da epidermólise bolhosa distrófica (DEB), antes conhecida como tipo Hallopeau-Siemens. Ela se caracteriza pela formação generalizada de bolhas e cicatrizes na pele e nas mucosas, associada a deformidades graves e ao comprometimento significativo de outros órgãos e sistemas.
Introdução
O que você precisa saber de cara
A epidermólise bolhosa distrófica recessiva grave e generalizada (RDEB-sev gen) é o subtipo mais grave da epidermólise bolhosa distrófica (DEB), antes conhecida como tipo Hallopeau-Siemens. Ela se caracteriza pela formação generalizada de bolhas e cicatrizes na pele e nas mucosas, associada a deformidades graves e ao comprometimento significativo de outros órgãos e sistemas.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 28 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 75 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisCleaves collagens of types I, II, and III at one site in the helical domain. Also cleaves collagens of types VII and X (PubMed:1645757, PubMed:2153297, PubMed:2557822). In case of HIV infection, interacts and cleaves the secreted viral Tat protein, leading to a decrease in neuronal Tat's mediated neurotoxicity (PubMed:16807369)
Secreted, extracellular space, extracellular matrix
Stratified squamous epithelial basement membrane protein that forms anchoring fibrils which may contribute to epithelial basement membrane organization and adherence by interacting with extracellular matrix (ECM) proteins such as type IV collagen
Secreted, extracellular space, extracellular matrix, basement membrane
Medicamentos aprovados (FDA)
1 medicamento encontrado nos registros da FDA americana.
Variantes genéticas (ClinVar)
1.232 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
15 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Epidermólise bolhosa distrófica generalizada autossômica recessiva, forma grave
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Novel variants impairing Sp1 transcription factor binding in the COL7A1 promoter cause mild cases of recessive dystrophic epidermolysis bullosa.
Recessive dystrophic epidermolysis bullosa (RDEB) is a rare and most often severe genodermatosis characterized by recurrent blistering and erosions of the skin and mucous membranes after minor trauma, leading to major local and systemic complications. RDEB is caused by loss-of-function mutations in COL7A1 encoding type VII collagen (C7), the main component of anchoring fibrils which form attachment structures stabilizing the cutaneous basement membrane zone. Most of the previously reported COL7A1 mutations are located in the coding or intronic regions. We describe 6 patients with localized or intermediate RDEB for whom one recessive pathogenic variant in the coding region and a second variant in the COL7A1 promoter were identified. These substitutions, three of which are novel, are localized in two Sp1 binding sites of the promoter region. DNA pull-down assay showed a drastic reduction of Sp1 binding consistent with a dramatic decrease in COL7A1 transcript and almost undetectable C7 protein levels. Our results reveal that mutations in the COL7A1 promoter on the background of a null allele can underlie localized or intermediate RDEB. They further emphasize the functional importance of Sp1 motifs in the proximal COL7A1 promoter which should be carefully investigated for regulatory mutations in the case of RDEB with only one pathogenic variant identified in the coding or intronic regions.
A pathogenic COL7A1 variant highlights semi-dominant inheritance in dystrophic epidermolysis bullosa.
Dystrophic epidermolysis bullosa is a rare subtype of inherited epidermolysis bullosa, caused by variants in the collagen type VII alpha 1 chain (COL7A1) gene (MIM120120). Both autosomal dominant and recessive inheritance has been reported with variable phenotype. We investigated a Pakistani family with dystrophic epidermolysis bullosa via exome sequencing and identified a pathogenic nonsense variant in COL7A1 NM_000094 c.1573 C > T:p.(Arg525*). The inheritance pattern observed was consistent with a semi-dominant model, where heterozygous parents exhibited a mild phenotype, and homozygous children were more severely affected. For dystrophic epidermolysis bullosa, loss-of-function variants are typically associated with the autosomal recessive form, while missense variants are linked to the autosomal dominant form. A review of the literature suggests a semi-dominance pattern for some missense variants, particularly glycine substitutions, but this concept had not been formally recognized. This study highlights the importance of considering semi-dominant inheritance models for dystrophic epidermolysis bullosa and other Mendelian diseases with an autosomal recessive mode of inheritance, as it can significantly impact diagnosis and genetic counseling.
Splice modulation strategy applied to deep intronic variants in COL7A1 causing recessive dystrophic epidermolysis bullosa.
Recessive dystrophic epidermolysis bullosa (RDEB) is a rare and most often severe genetic disease characterized by recurrent blistering and erosions of the skin and mucous membranes after minor trauma, leading to major local and systemic complications. The disease is caused by loss-of-function variants in COL7A1 encoding type VII collagen (C7), the main component of anchoring fibrils, which form attachment structures stabilizing the cutaneous basement membrane zone. Alterations in C7 protein structure and/or expression lead to abnormal, rare or absent anchoring fibrils resulting in loss of dermal-epidermal adherence and skin blistering. To date, more than 1,200 distinct COL7A1 deleterious variants have been reported and 19% are splice variants. Here, we describe two RDEB patients for whom we identified two pathogenic deep intronic pathogenic variants in COL7A1. One of these variants (c.7795-97C > G) promotes the inclusion of a pseudoexon between exons 104 and 105 in the COL7A1 transcript, while the other causes partial or complete retention of intron 51. We used antisense oligonucleotide (ASO) mediated exon skipping to correct these aberrant splicing events in vitro. This led to increased normal mRNA splicing above 94% and restoration of C7 protein expression at a level (up to 56%) that should be sufficient to reverse the phenotype. This first report of exon skipping applied to counteract deep intronic variants in COL7A1 represents a promising therapeutic strategy for personalized medicine directed at patients with intronic variants at a distance of consensus splice sites.
Epidermolysis Bullosa: Two rare case reports of COL7A1 and EBS-GEN SEV KRT14 variants with review of literature.
Bullosa is a rare hereditary skin condition that causes blisters. Genes encoding structural proteins at or near the dermal-epidermal junction are mutated recessively or dominantly, and this is the primary cause of EB. Herein, two Chinese boys were diagnosed with the condition, each with a different variant in a gene that serves as a reference for EB genetic counseling. Skincare significantly impacted their prognosis and quality of life. Two Chinese boys, with phenotypically normal parents, have been diagnosed with distinct blister symptoms, one with Dominant Dystrophic Epidermolysis Bullosa and the other with a severe form of Epidermolysis Bullosa Simplex. The first patient had a G-to-A variant in the COL7A1 allele, at nucleotide position 6163 which was named "G2055A". The proband is heterozygous for Dystrophic Epidermolysis Bullosa due to a COL7A1 allele with a glycine substitution at the triple helix domain. A similar variant has been discovered in his mother, indicating its potential transmission to future generations. Another patient had severe Epidermolysis Bullosa Simplex with a rare c.377T > A variant resulting in substitution of amino acid p.Leu126Arg (NM_000526.5 (c.377T > G, p.Leu126Arg) in the Keratin 14 gene. In prior literature, Keratin 14 has been associated with an excellent prognosis. However, our patient with this infrequent variant tragically died from sepsis at 21 days old. There has been a reported occurrence of the variant only once. Our study reveals that Epidermolysis Bullosa patients with COL7A1 c.6163G > A and KRT14 c.377T>A variants have different clinical presentations, with dominant forms of Dystrophic EB having milder phenotypes than recessive ones. Thus, the better prognosis in the c.6163G > A patient. Furthermore, c.377T>A patient was more prone to infection than the patient with c.6163G>A gene variant. Genetic testing is crucial for identifying the specific variant responsible and improving treatment options.
Splicing Modulation via Antisense Oligonucleotides in Recessive Dystrophic Epidermolysis Bullosa.
Antisense oligonucleotides (ASOs) represent an emerging therapeutic platform for targeting genetic diseases by influencing various aspects of (pre-)mRNA biology, such as splicing, stability, and translation. In this study, we investigated the potential of modulating the splicing pattern in recessive dystrophic epidermolysis bullosa (RDEB) patient cells carrying a frequent genomic variant (c.425A > G) that disrupts splicing in the COL7A1 gene by using short 2'-O-(2-Methoxyethyl) oligoribo-nucleotides (2'-MOE ASOs). COL7A1-encoded type VII collagen (C7) forms the anchoring fibrils within the skin that are essential for the attachment of the epidermis to the underlying dermis. As such, gene variants of COL7A1 leading to functionally impaired or absent C7 manifest in the form of extensive blistering and wounding. The severity of the disease pattern warrants the development of novel therapies for patients. The c.425A > G variant at the COL7A1 exon 3/intron 3 junction lowers the efficiency of splicing at this junction, resulting in non-functional C7 transcripts. However, we found that correct splicing still occurs, albeit at a very low level, highlighting an opportunity for intervention by modulating the splicing reaction. We therefore screened 2'-MOE ASOs that bind along the COL7A1 target region ranging from exon 3 to the intron 3/exon 4 junction for their ability to modulate splicing. We identified ASOs capable of increasing the relative levels of correctly spliced COL7A1 transcripts by RT-PCR, sqRT-PCR, and ddPCR. Furthermore, RDEB-derived skin equivalents treated with one of the most promising ASOs exhibited an increase in full-length C7 expression and its accurate deposition along the basement membrane zone (BMZ).
📚 EuropePMCmostrando 28
A pathogenic COL7A1 variant highlights semi-dominant inheritance in dystrophic epidermolysis bullosa.
BMC medical genomicsNovel variants impairing Sp1 transcription factor binding in the COL7A1 promoter cause mild cases of recessive dystrophic epidermolysis bullosa.
European journal of human genetics : EJHGA homozygous nonsense mutation identified in COL7A1 in a family with autosomal recessive dystrophic epidermolysis bullosa.
Journal of medicine and lifeSplice modulation strategy applied to deep intronic variants in COL7A1 causing recessive dystrophic epidermolysis bullosa.
Proceedings of the National Academy of Sciences of the United States of AmericaUnravelling drivers of cutaneous squamous cell carcinoma in recessive dystrophic epidermolysis bullosa.
Human immunologyEpidermolysis Bullosa: Two rare case reports of COL7A1 and EBS-GEN SEV KRT14 variants with review of literature.
BMC pediatricsElevated expression of interleukin-6 (IL-6) and serum amyloid A (SAA) in the skin and the serum of recessive dystrophic epidermolysis bullosa: Skin as a possible source of IL-6 through Toll-like receptor ligands and SAA.
Experimental dermatologySplicing Modulation via Antisense Oligonucleotides in Recessive Dystrophic Epidermolysis Bullosa.
International journal of molecular sciencesHighly Efficient Ex Vivo Correction of COL7A1 through Ribonucleoprotein-Based CRISPR/Cas9 and Homology-Directed Repair to Treat Recessive Dystrophic Epidermolysis Bullosa.
The Journal of investigative dermatologyIdentification of a novel COL7A1 variant associated with dystrophic epidermolysis bullosa pruriginosa responding effectively to dupilumab.
Molecular genetics & genomic medicineCOL7A1 Editing via RNA Trans-Splicing in RDEB-Derived Skin Equivalents.
International journal of molecular sciencesOur experience of using Losartan for esophageal stenosis in children with dystrophic form of congenital epidermolysis bullosa.
Journal of pediatric surgeryIleus as a complication of butterfly wing disease case report.
Rozhledy v chirurgii : mesicnik Ceskoslovenske chirurgicke spolecnostiIn Vitro Models for the Evaluation of Antisense Oligonucleotides in Skin.
Methods in molecular biology (Clifton, N.J.)A Case Report of an Infant with Autosomal Recessive Dystrophic Epidermolysis Bullosa: COL7A1 Gene Mutations at C2005T and G7922A.
Acta dermatovenerologica Croatica : ADCFuture applications of 3D bioprinting: A promising technology for treating recessive dystrophic epidermolysis bullosa.
Experimental dermatologyDoes fludarabine have a beneficial effect in recessive dystrophic epidermolysis bullosa?
The British journal of dermatologyDiversity of Mechanisms Underlying Latent TGF-β Activation in Recessive Dystrophic Epidermolysis Bullosa.
The Journal of investigative dermatologyApparent Missense Variant in COL7A1 Causes a Severe Form of Recessive Dystrophic Epidermolysis Bullosa via Effects on Splicing.
Acta dermato-venereologicaPredictable CRISPR/Cas9-Mediated COL7A1 Reframing for Dystrophic Epidermolysis Bullosa.
The Journal of investigative dermatologySafety and early efficacy outcomes for lentiviral fibroblast gene therapy in recessive dystrophic epidermolysis bullosa.
JCI insightIdentification of Rigosertib for the Treatment of Recessive Dystrophic Epidermolysis Bullosa-Associated Squamous Cell Carcinoma.
Clinical cancer research : an official journal of the American Association for Cancer ResearchMurine type VII collagen distorts outcome in human skin graft mouse model for dystrophic epidermolysis bullosa.
Experimental dermatologyClinical algorithm to manage anemia in epidermolysis bullosa.
Pediatric dermatologyA rare case of skin blistering and esophageal stenosis in the course of epidermolysis bullosa - case report and literature review.
BMC gastroenterologyIL-6/IL-10 Ratio as A Prognostic and Predictive Marker of the Severity of Inherited Epidermolysis Bullosa.
Iranian journal of immunology : IJIAn RNA-targeted therapy for dystrophic epidermolysis bullosa.
Nucleic acids researchFeasibility, efficacy, and safety of ultrasound-guided axillary plexus blockade in pediatric patients with epidermolysis bullosa dystrophica.
Paediatric anaesthesiaAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Epidermólise bolhosa distrófica generalizada autossômica recessiva, forma grave.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Epidermólise bolhosa distrófica generalizada autossômica recessiva, forma grave
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Novel variants impairing Sp1 transcription factor binding in the COL7A1 promoter cause mild cases of recessive dystrophic epidermolysis bullosa.
- A pathogenic COL7A1 variant highlights semi-dominant inheritance in dystrophic epidermolysis bullosa.
- Splice modulation strategy applied to deep intronic variants in COL7A1 causing recessive dystrophic epidermolysis bullosa.Proceedings of the National Academy of Sciences of the United States of America· 2024· PMID 39159368mais citado
- Epidermolysis Bullosa: Two rare case reports of COL7A1 and EBS-GEN SEV KRT14 variants with review of literature.
- Splicing Modulation via Antisense Oligonucleotides in Recessive Dystrophic Epidermolysis Bullosa.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:79408(Orphanet)
- OMIM OMIM:226600(OMIM)
- MONDO:0009179(MONDO)
- GARD:6308(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Q7302398(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
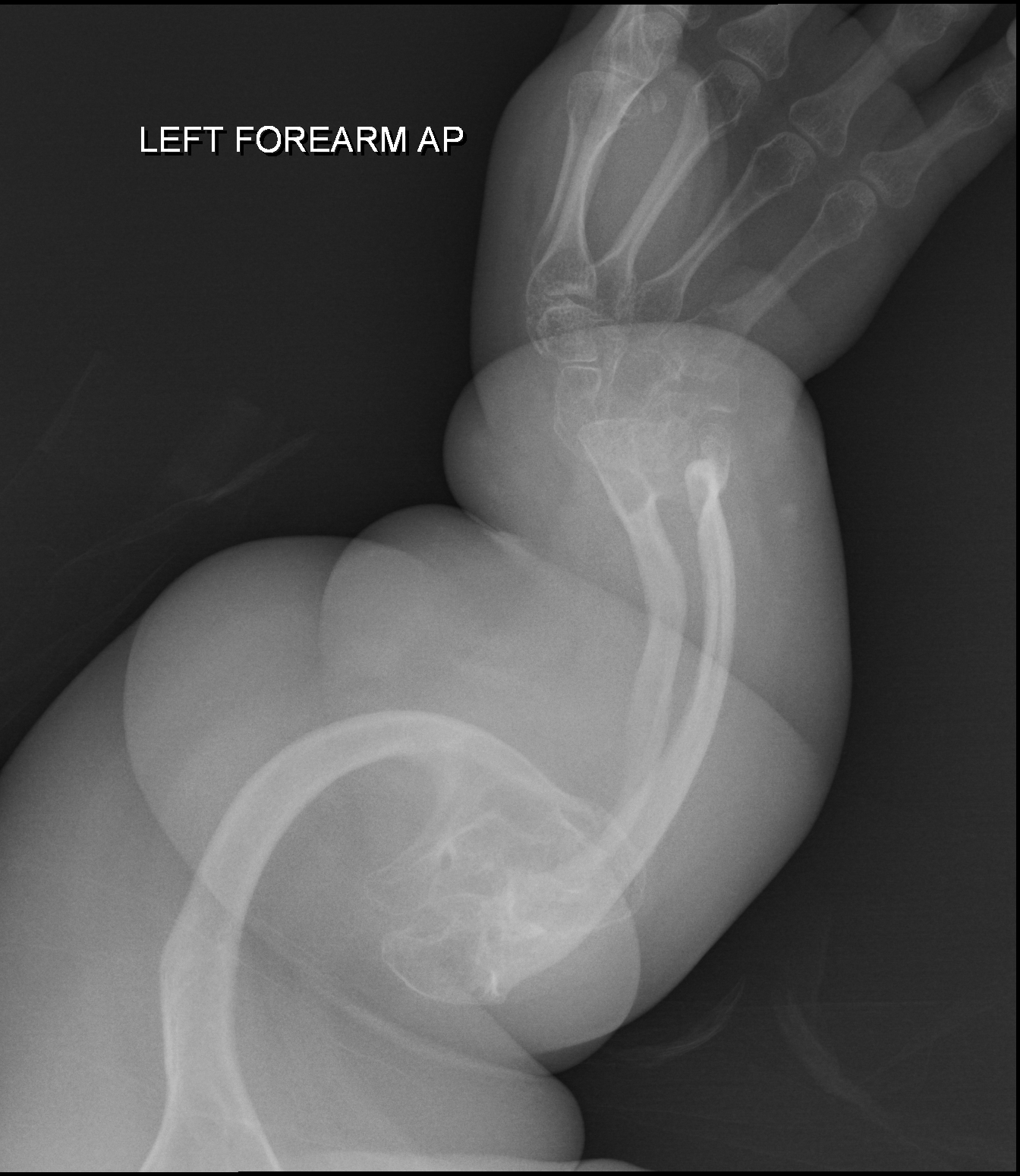
Epidermólise bolhosa distrófica generalizada autossômica recessiva, forma grave
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos aprovados FDA
- fonte: FDA OpenFDA