A Puberdade Precoce Familiar Limitada a Meninos (FMPP) é uma condição familiar de puberdade precoce que afeta apenas meninos e não é causada pelos hormônios cerebrais que normalmente iniciam esse processo. Geralmente, ela se manifesta entre 2 e 5 anos de idade, com um crescimento acelerado, o desenvolvimento precoce de características sexuais secundárias (como pelos e engrossamento da voz) e uma altura final menor na vida adulta.
Introdução
O que você precisa saber de cara
A Puberdade Precoce Familiar Limitada a Meninos (FMPP) é uma condição familiar de puberdade precoce que afeta apenas meninos e não é causada pelos hormônios cerebrais que normalmente iniciam esse processo. Geralmente, ela se manifesta entre 2 e 5 anos de idade, com um crescimento acelerado, o desenvolvimento precoce de características sexuais secundárias (como pelos e engrossamento da voz) e uma altura final menor na vida adulta.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 7 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 14 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisReceptor for lutropin-choriogonadotropic hormone (PubMed:11847099). The activity of this receptor is mediated by G proteins which activate adenylate cyclase (PubMed:11847099)
Cell membrane
Familial male precocious puberty
In FMPP the receptor is constitutively activated.
Variantes genéticas (ClinVar)
89 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Puberdade precoce familiar limitada ao sexo masculino
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Precocious puberty: An overview of pathogenesis, clinical presentation, and management.
Precocious puberty, defined as the onset of secondary sexual characteristics at 2.5 SD below the mean age of onset of puberty (8 years in girls and 9 years in boys) is a complex clinical condition that has considerable physiological and psychological consequences on children and their parents. There are two types - central (gonadotrophin-dependent) and peripheral (gonadotrophin independent). Among the causes of central precocious puberty are various genetic mutations, syndromes and central nervous system disorders that prematurely activate the hypothalamic pituitary gonadal axis leading to the secretion of sex steroids that induced pubertal changes. Peripheral precocious puberty is commonly secondary to isolated sources of sex steroids such as tumours, exogenous steroids or as part of the McCune Albright syndrome. Long-term consequences of precocious puberty especially the central type include short stature and psychological problems. Children presenting with features of precocious puberty (PP) must be thoroughly assessed starting with a detailed history, physical examination and initiation of appropriate investigations followed by categorisation of the PP. A multidisciplinary team (consisting of a paediatrician an adolescent gynaecologist, a paediatric endocrinologist, a geneticist and a clinical psychologist) is essential for management. Treatment should aim at arresting or reversing the pubertal changes, counselling and support both for the children and their families and, addressing the implications for genetic causes for the family (if any). Untreated, precocious puberty may have considerable negative psychological and medical impact on the child. Central precocious puberty (CPP) is defined as the early activation of the hypothalamic-pituitary-gonadal (HPG) axis, occurring before age 8 in girls and 9 in boys and leading to the early appearance of secondary sexual characteristics. While multiple environmental and pathological factors may contribute to the condition, genetic and epigenetic mechanisms have emerged as major determinants in both familial and sporadic cases. This chapter reviews current knowledge on the genetic architecture of CPP, including well-established monogenic causes such as mutations in MKRN3, DLK1, KISS1, KISS1R and MECP2, and other candidate genes. The role of genomic imprinting is discussed in detail. In addition, recent insights into epigenetic regulation—including differential methylation patterns in hypothalamic genes and the impact of imprinted gene networks—highlight emerging mechanisms that may bridge genetic susceptibility with environmental influences. The chapter also explores syndromic forms of CPP and cases in which CPP arises secondarily to other genetic disorders. These include children with neurodevelopmental disabilities and those with congenital adrenal hyperplasia (CAH), in which chronic peripheral androgen excess may lead to premature activation of the central HPG axis. Together, this chapter provides a comprehensive and up-to-date overview of the molecular and genetic mechanisms underlying CPP, offering clinicians and researchers a framework for understanding its pathogenesis, refining diagnostic approaches, and identifying potential targets for future therapies. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG. Puberty is a complex developmental transition marked by the appearance of secondary sexual characteristics, accelerated growth, and profound physical and psychological changes that culminate in reproductive maturity. A combination of intrinsic factors, such as genetics, and extrinsic factors, including environmental and nutritional influences, plays a role in the onset and progression of puberty. Precocious puberty is defined as the onset of secondary sexual characteristics before age 8 in girls and before age 9 in boys. This definition, endorsed by major pediatric and endocrine societies, remains the standard threshold for initiating a diagnostic evaluation. Although about 3% of children fall below this statistical threshold, showing pubertal signs more than 2 standard deviations earlier than the median age, most cases are benign variants, such as premature thelarche or premature adrenarche, that are self-limited, nonprogressive, and need no treatment. Assessing puberty's clinical context and progression is essential for distinguishing true precocious puberty from normal variants. There are 2 main types of precocious puberty: central and peripheral. Central precocious puberty results from the premature activation of the hypothalamic–pituitary–gonadal axis, leading to gonadotropin-dependent sexual maturation. Peripheral precocious puberty is gonadotropin-independent and driven by excess sex steroid production from the gonads, adrenals, or exogenous sources. True precocious puberty affects approximately 0.2% of girls and fewer than 0.05% of boys. In recent decades, puberty has started earlier, especially in girls, likely due to increasing childhood obesity and environmental factors. Untreated precocious puberty can lead to psychosocial issues and reduced adult height due to early epiphyseal closure. Moreover, failing to diagnose precocious puberty promptly may result in the missed detection of severe underlying conditions that require urgent medical or surgical intervention. Differentiating harmless normal variants from true precocious puberty can be challenging, especially in borderline cases. Effective treatment for central precocious puberty is available with gonadotropin-releasing hormone analogs, while options for peripheral precocious puberty are more limited and often less effective. A careful, family-centered approach is essential to guide personalized management decisions in these cases.
Peripheral Precocious Puberty Due to Autonomous Gonadal Activation: A Multicenter Experience.
Peripheral precocious puberty (PP) is far less commonly encountered compared to central precocious puberty (CPP) in pediatric endocrine practice. Long-standing non-diagnosis may cause rapidly advancing bone age, leading to CPP and resultant short stature. We aimed to report the clinical profile and current Indian experience in the management of children with peripheral PP due to autonomous gonadal activation. This multicentric retrospective study reports data on 23 children (20 girls) presenting with peripheral PP as a result of autonomous gonadal activation from eight pediatric endocrine centers across India. Their clinicodemographic, anthropometric, and laboratory measurements were reviewed. The mean ± SD chronological age at the time of presentation was 4.9 ± 2.0 years, and the mean bone age was 7.6 ± 2.6 years. Nine (39%) children were tall for mid-parental height. Thirteen (57%) children were diagnosed with McCune Albright syndrome (MAS), one boy (4%) with familial male-limited PP, and nine (39%) with functional ovarian cysts. Ninety-five percent of girls had menarche as their presenting complaint, with mean age at menarche being 4.6 ± 2.3 years. Ovarian cysts were present in 16 girls, of whom seven (43.8%) had MAS. PP in seven (30%) children progressed to CPP, with mean age of CPP being 6.6 ± 2.1 years. Letrozole was the primary drug of choice, while leuprolide acetate was added in children who progressed to CPP. We report the clinical profile of children with peripheral PP due to autonomous gonadal activation. Clinical diagnosis and timely intervention to halt the progression of puberty and prevent early epiphyseal fusion, thereby improving final height, along with close follow-up, are vital in the management of these disorders.
The clinical characteristics of 10 cases and adult height of six cases of rare familial male-limited precocious puberty.
Familial Male-Limited Precocious Puberty (FMPP) is a rare autosomal-dominant genetic condition with sexual dimorphism. We aim to summarize the clinical characteristics of FMPP patients and emphasize the use of a therapeutic regimen involving letrozole, spironolactone, and GnRHa, to augment clinician's understanding of the disease, thus enhancing patient care. We retrospectively analyzed the clinical data of 10 FMPP patients and conducted follow-up assessments of adult height in six patients. Out of the 10 FMPP cases, five had the LHCGR M398T mutation, three exhibited the LHCGR A564G mutation, and two had the LHCGR T577I mutation. All patients initially presented with symptoms like penile enlargement, frequent erections, and rapid growth. Their median age at diagnosis was 4.67 years with bone age being 9 years. Four patients were untreated with a median adult height of 162 cm. Six patients underwent treatment between ages 3.58 and 5.5 years noting decreased frequency of erections, slower growth rate, and delayed bone age progression. Secondary Central Precocious Puberty (CPP) developed between ages 5 and 6.5 years in all cases, necessitating additional GnRHa treatment. Two treated cases reached an adult height of 176 cm and 173 cm, respectively, without any significant adverse effects. The most prevalent genotype among FMPP patients in this study was the LHCGR M398T mutation. Early intervention using a regimen including letrozole and spironolactone, and later GnRHa, appears beneficial in limiting physical signs and improving adult height without major side effects. However, the longer-term effects on fertility require further investigation.
Lipidomics reveals ceramide biomarkers for detecting central precocious puberty in girls.
Pubertal timing is modulated by complex interactions between the pituitary and gonadal sex steroid hormones. Evidence indicates that sphingolipids are involved in the biosynthesis of steroid hormones at multiple levels. This study recruited adolescent female patients from pubertal and pediatric endocrine clinics in Northern and Southern Taiwan from the Taiwan Puberty Longitudinal Study. A total of 112 plasma samples (22 healthy control, 29 peripheral precocious puberty (PPP), and 61 CPP samples) were collected. We extracted lipids from the plasma samples using the modified Folch method. The un-targeted ultrahigh-performance liquid chromatography tandem mass spectrometry (UPLC-MS/MS) was employed for the lipid analysis. We identified sphingolipid-linked metabolites, including Cer(18:0/15:0), Cer(18:1/16:0), and Cer(18:1/26:0) as candidate biomarkers for distinguishing girls with CPP from the control group by using an excellent discrimination model (AUC = 0.964). Moreover, Cer(18:0/22:0) and Cer(d18:0/18:1) were identified as potential biomarkers of PPP, with an AUC value of 0.938. Furthermore, CerP(18:1/18:0) was identified as the sole candidate biomarker capable of differentiating CPP from PPP. The biomarkers identified in this study can facilitate the accurate detection of CPP in girls, provide insights into lipid-linked pathophysiology, and present a novel method of monitoring the progression of this disorder.
Presentation and Care for Children with Peripheral Precocious Puberty.
Peripheral precocious puberty (PPP) refers to the early onset of sexual maturation that is independent of central nervous system control. The extensive differential diagnosis includes congenital and acquired causes. Presenting features depend on which class of sex steroids is involved, and diagnosis rests on hormonal and, if indicated, imaging and/or genetic studies. Effective treatment exists for nearly all causes of PPP. Ongoing research will advance our therapeutic armamentarium and understanding of the pathophysiologic basis of these conditions.
Publicações recentes
Peripheral Precocious Puberty Due to Autonomous Gonadal Activation: A Multicenter Experience.
The clinical characteristics of 10 cases and adult height of six cases of rare familial male-limited precocious puberty.
Presentation and Care for Children with Peripheral Precocious Puberty.
Familial male-limited precocious puberty due to an activating mutation of the LHCGR: a case report and literature review.
Precocious Puberty: Types, Pathogenesis and Updated Management.
📚 EuropePMCmostrando 39
Precocious puberty: An overview of pathogenesis, clinical presentation, and management.
Best practice & research. Clinical obstetrics & gynaecologyPeripheral Precocious Puberty Due to Autonomous Gonadal Activation: A Multicenter Experience.
CureusThe clinical characteristics of 10 cases and adult height of six cases of rare familial male-limited precocious puberty.
Journal of pediatric endocrinology & metabolism : JPEMLipidomics reveals ceramide biomarkers for detecting central precocious puberty in girls.
Obesity research & clinical practicePresentation and Care for Children with Peripheral Precocious Puberty.
Endocrinology and metabolism clinics of North AmericaDiagnosis of Central Precocious Puberty.
Endocrinology and metabolism clinics of North AmericaPrecocious Puberty: Types, Pathogenesis and Updated Management.
CureusCentral precocious puberty secondary to peripheral precocious puberty due to a pineal germ cell tumor: a case and review of literature.
BMC endocrine disordersClinical course of peripheral precocious puberty in girls due to autonomous ovarian cysts.
Clinical endocrinologyRare variants in the MECP2 gene in girls with central precocious puberty: a translational cohort study.
The lancet. Diabetes & endocrinology[Further progress of the etiology,diagnosis and treatment of peripheral precocious puberty].
Zhonghua yu fang yi xue za zhi [Chinese journal of preventive medicine]Association between Serum Per- and Polyfluoroalkyl Substance Levels and Risk of Central and Peripheral Precocious Puberty in Girls.
Environmental science & technologyPeripheral precocious puberty in Li-Fraumeni syndrome: a case report and literature review of pure androgen-secreting adrenocortical tumors.
Journal of medical case reportsHuman chorionic gonadotrophin secreting adrenocortical neoplasm presenting with peripheral precocious puberty in an infant.
Journal of pediatric endocrinology & metabolism : JPEMFamilial Male-limited Precocious Puberty (FMPP) and Testicular Germ Cell Tumors.
The Journal of clinical endocrinology and metabolismRapidly Progressive Precocious Puberty With an Elevated Testosterone Level in a 5-Year-Old Boy With a β-Human Chorionic Gonadotropin-Secreting Intracranial Germ Cell Tumor in the Pineal Gland.
AACE clinical case reportsLong-Term Treatment With Letrozole in a Boy With Familial Male-Limited Precocious Puberty.
Frontiers in endocrinologyPeripheral Precocious Puberty of Ovarian Origin in a Series of 18 Girls: Exome Study Finds Variants in Genes Responsible for Hypogonadotropic Hypogonadism.
Frontiers in pediatricsPrecocious Puberty in Boys: A Study Based on Five Years of Data from a Single Center in Northern China.
Journal of clinical research in pediatric endocrinologyDiagnosis and management of precocious sexual maturation: an updated review.
European journal of pediatricsCongenital adrenal hyperplasia due to 11-Beta-hydroxylase deficiency in a Tunisian family.
The Pan African medical journalGrowing Up Fast: Managing Autism Spectrum Disorder and Precocious Puberty.
Journal of developmental and behavioral pediatrics : JDBPDelayed and Precocious Puberty: Genetic Underpinnings and Treatments.
Endocrinology and metabolism clinics of North AmericaIsolated premature menarche in two siblings with Neurofibromatosis type 1.
Journal of pediatric endocrinology & metabolism : JPEMEstrogen can promote the expression of genes related to precocious puberty in GT1-7 mouse hypothalamic GnRH neuronal cell line via activating G protein-coupled estrogen receptor.
General physiology and biophysicsTestotoxicosis without Testicular Mass: Revealed by Peripheral Precocious Puberty and Confirmed by Somatic LHCGR Gene Mutation.
Endocrine researchPeripheral precocious puberty including congenital adrenal hyperplasia: causes, consequences, management and outcomes.
Best practice & research. Clinical endocrinology & metabolismNovel DNA variation of GPR54 gene in familial central precocious puberty.
Italian journal of pediatricsA Case of Familial Male-Limited Precocious Puberty in a Child With Klinefelter Syndrome.
Journal of the Endocrine SocietyMKRN3 Levels in Girls with Central Precocious Puberty during GnRHa Treatment: A Longitudinal Study.
Hormone research in paediatricsCentral Precocious Puberty Caused by a Heterozygous Deletion in the MKRN3 Promoter Region.
Neuroendocrinology[Familial male-limited precocious puberty due to Asp578His mutations in the LHCGR gene: clinical characteristics and gene analysis in an infant].
Zhongguo dang dai er ke za zhi = Chinese journal of contemporary pediatricsGerm Cell Neoplasia in Situ and Preserved Fertility Despite Suppressed Gonadotropins in a Patient With Testotoxicosis.
The Journal of clinical endocrinology and metabolismMKRN3 levels in girls with central precocious puberty and correlation with sexual hormone levels: a pilot study.
EndocrineA novel DAX-1 mutation in two male siblings presenting with precocious puberty and late-onset hypogonadotropic hypogonadism.
Journal of pediatric endocrinology & metabolism : JPEMTestotoxicosis: Report of Two Cases, One with a Novel Mutation in LHCGR Gene.
Journal of clinical research in pediatric endocrinologyTreatment of Peripheral Precocious Puberty.
Endocrine developmentSexual Precocity--Genetic Bases of Central Precocious Puberty and Autonomous Gonadal Activation.
Endocrine developmentPrecocious puberty in children.
Journal of the College of Physicians and Surgeons--Pakistan : JCPSPAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Puberdade precoce familiar limitada ao sexo masculino.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Puberdade precoce familiar limitada ao sexo masculino
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Precocious puberty: An overview of pathogenesis, clinical presentation, and management.
- Peripheral Precocious Puberty Due to Autonomous Gonadal Activation: A Multicenter Experience.
- The clinical characteristics of 10 cases and adult height of six cases of rare familial male-limited precocious puberty.
- Lipidomics reveals ceramide biomarkers for detecting central precocious puberty in girls.
- Presentation and Care for Children with Peripheral Precocious Puberty.
- Familial male-limited precocious puberty due to an activating mutation of the LHCGR: a case report and literature review.
- Precocious Puberty: Types, Pathogenesis and Updated Management.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:3000(Orphanet)
- OMIM OMIM:176410(OMIM)
- MONDO:0008303(MONDO)
- GARD:4475(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q5432947(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
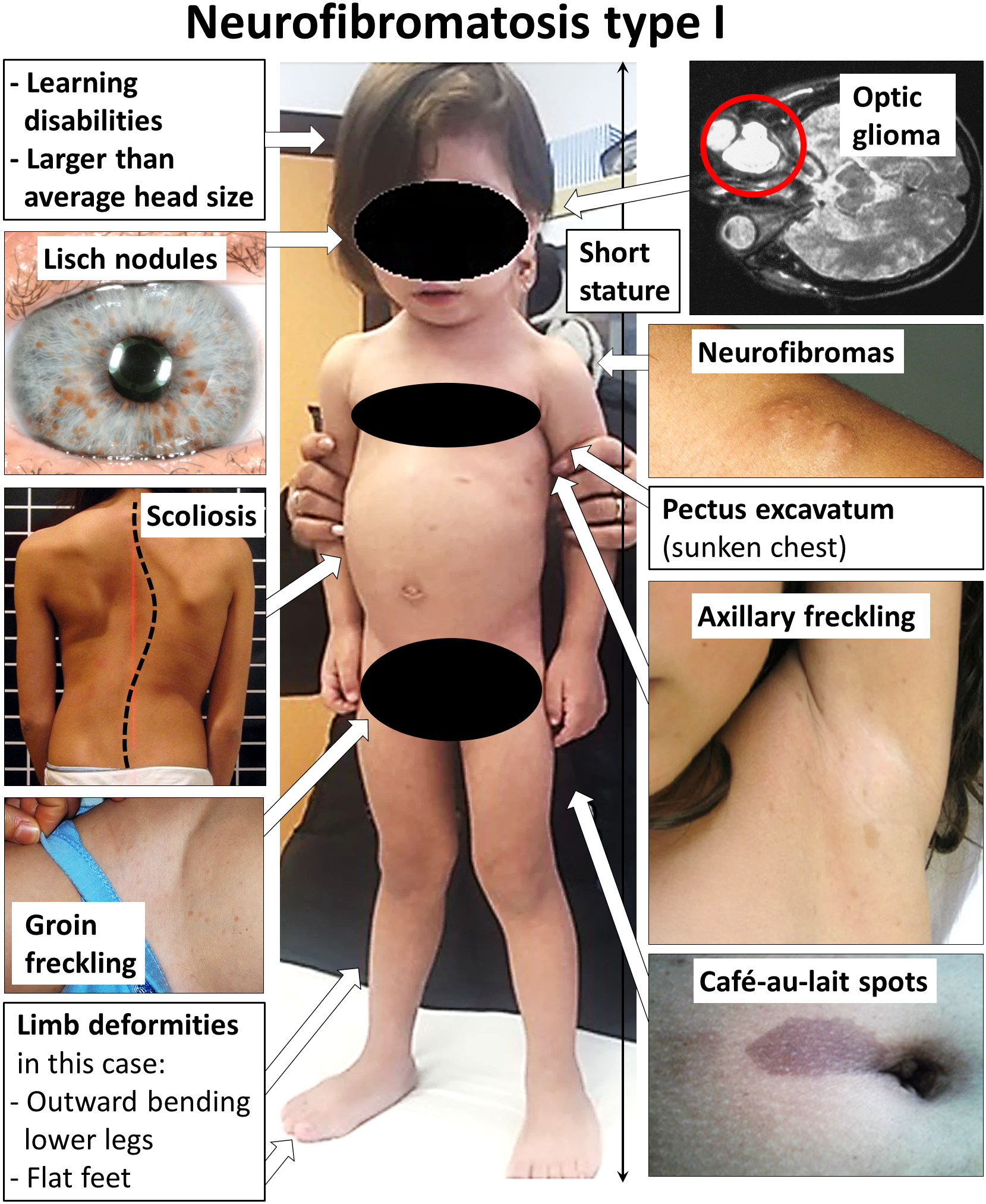
Puberdade precoce familiar limitada ao sexo masculino
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Reposicionamento
- fonte: Drug Repurposing Hub