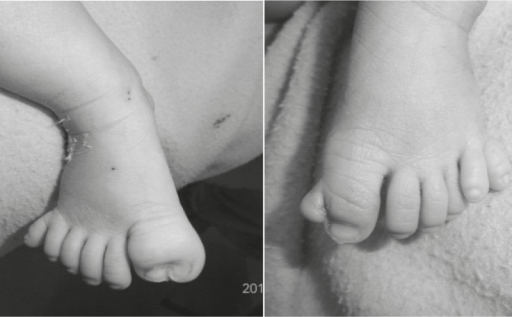
A síndrome acrocalosa (SCA) é uma síndrome polimalformativa caracterizada por agenesia do corpo caloso (CC), anomalias distais dos membros, anomalias craniofaciais menores e déficit intelectual.
Introdução
O que você precisa saber de cara
A síndrome acrocalosa (SCA) é uma síndrome polimalformativa caracterizada por agenesia do corpo caloso (CC), anomalias distais dos membros, anomalias craniofaciais menores e déficit intelectual.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 28 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 90 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisEssential for hedgehog signaling regulation: acts both as a negative and positive regulator of sonic hedgehog (Shh) and Indian hedgehog (Ihh) pathways, acting downstream of SMO, through both SUFU-dependent and -independent mechanisms (PubMed:21633164). Involved in the regulation of microtubular dynamics. Required for proper organization of the ciliary tip and control of ciliary localization of SUFU-GLI2 complexes (By similarity). Required for localization of GLI3 to cilia in response to Shh. Neg
Cell projection, ciliumCytoplasm, cytoskeleton, cilium basal body
Has a dual function as a transcriptional activator and a repressor of the sonic hedgehog (Shh) pathway, and plays a role in limb development. The full-length GLI3 form (GLI3FL) after phosphorylation and nuclear translocation, acts as an activator (GLI3A) while GLI3R, its C-terminally truncated form, acts as a repressor. A proper balance between the GLI3 activator and the repressor GLI3R, rather than the repressor gradient itself or the activator/repressor ratio gradient, specifies limb digit num
NucleusCytoplasmCell projection, cilium
Greig cephalo-poly-syndactyly syndrome
Autosomal dominant disorder affecting limb and craniofacial development. It is characterized by pre- and postaxial polydactyly, syndactyly of fingers and toes, macrocephaly and hypertelorism.
Variantes genéticas (ClinVar)
650 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 1.475 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
5 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome acrocalosal
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Midline defect with corpus callosum agenesis, vermian hypoplasia and median cleft lip palate.
Midline defects in the brain may be related to genetic syndromes. Association with facial anomalies and skeletal deformities has been described. In the present case, a routine second trimester scan revealed cerebral abnormalities (corpus callosum agenesis, cerebellar cleft due to vermian hypoplasia, ventriculomegaly), suspected cortical developmental disorder, hypertelorism, a hypoplastic nasal bone, a small median cleft lip and palate, abnormal facial profile, as well as syndactyly of the left hand involving the fourth and fifth finger. Genetic testing revealed a normal karyotype. Subsequent trio exome sequencing did not identify any pathogenic variants or variants of unknown significance. The vaginal delivery at term and postnatal adaptation were uneventful. Postnatal neurosonographic imaging and clinical evaluation confirmed the prenatal findings. Both mother and child were discharged in healthy condition with scheduled follow-ups. Differential diagnoses of the present anomalies include Hartsfield-Bixler-Demyer Syndrome, Oro-Facial-Digital-Syndromes, Ectrodactyly Ectodermal Dysplasia Cleft Lip/Palate Syndrome and Acrocallosal Syndrome. Invasive diagnostic and genetic testing are recommended when multiple fetal anomalies suggest a potential genetic syndrome. While not all cases reveal an underlying genetic cause, prenatal findings can provide valuable information to help parents and healthcare providers make informed decisions about the continuation of the pregnancy.
Retinitis Pigmentosa Sine Pigmento in a Patient With a Heterozygous Mutation on the KIF7 Gene: A Case Report.
Mutations in the KIF7 gene have been implicated in autosomal recessive conditions such as Joubert syndrome, acrocallosal syndrome, and fetal hydrolethalus, as well as in retinal degeneration and other ocular manifestations due to their effect on primary cilia. In this study, we report that the full-field electroretinogram (ERG) test showed non-recordable scotopic ERG responses, while photopic ERG responses were diminished bilaterally. This is a case report of a 62-year-old female patient with painless, progressive vision loss in both eyes. Fundus examination revealed a pale optic nerve head, vessel attenuation, and macular thinning without peripheral pigmentary changes. The full-field electroretinogram (ERG) test showed non-recordable scotopic ERG responses, while photopic ERG responses were diminished bilaterally. Based on these ocular findings, the patient was clinically diagnosed with retinitis pigmentosa (RP) sine pigmento. Genetic testing identified a pathogenic heterozygous mutation in the KIF7 gene with the variant c.61C>T (p.Arg21*). Our case suggests that this pathologic variant may be associated with RP sine pigmento. Further studies are warranted to better understand the role of the KIF7 gene in retinal dystrophies.
Intracranial arachnoid cysts.
The purpose of this review article is to outline the natural history, pathogenesis, anatomic considerations and surgical decision-making in caring for patients with intracranial arachnoid cysts. A review of the literature for intracranial arachnoid cysts was performed using Embase, PubMed, and Web of Science databases, including review of the bibliographies of eligible articles and the author's own experience. Among those reviewed, 59 relevant original articles were included as well as illustrative cases from the authors own experience. Arachnoid cysts are congenital lesions characterized by split arachnoid membrane, thick collagen in the cyst wall, absent traversing trabecular processes within the cyst, and hyperplastic arachnoid cells in the cyst wall. The underlying etiology is not entirely known, and they occur in greater proportion in males and in greater incidence with various genetic conditions including Down syndrome, mucopolysaccharidosis, schizencephaly, neurofibromatosis, autosomal dominant polycystic kidney disease (ADPKD), acrocallosal syndrome, and Aicardi syndrome. Most intracranial arachnoid cysts are incidentally found and occur in the middle cranial fossa, with the remaining occurring in the cerebellopontine angle, suprasellar cistern, quadrigeminal cistern, convexity, and posterior fossa/cisterna magna. The current article outlines the natural history, prevalence, demographic factors, and treatment decisions in managing patients with intracranial arachnoid cysts.
Familial and syndromic forms of arachnoid cyst implicate genetic factors in disease pathogenesis.
Arachnoid cysts (ACs) are the most common space-occupying lesions in the human brain and present significant challenges for clinical management. While most cases of ACs are sporadic, nearly 40 familial forms have been reported. Moreover, ACs are seen with increased frequency in multiple Mendelian syndromes, including Chudley-McCullough syndrome, acrocallosal syndrome, and autosomal recessive primary ciliary dyskinesia. These findings suggest that genetic factors contribute to AC pathogenesis. However, traditional linkage and segregation approaches have been limited in their ability to identify causative genes for ACs because the disease is genetically heterogeneous and often presents asymptomatically and sporadically. Here, we comprehensively review theories of AC pathogenesis, the genetic evidence for AC formation, and discuss a different approach to AC genomics that could help elucidate this perplexing lesion and shed light on the associated neurodevelopmental phenotypes seen in a significant subset of these patients.
Sequences in the stalk domain regulate auto-inhibition and ciliary tip localization of the immotile kinesin-4 KIF7.
The kinesin-4 member KIF7 plays critical roles in Hedgehog signaling in vertebrate cells. KIF7 is an atypical kinesin as it binds to microtubules but is immotile. We demonstrate that, like conventional kinesins, KIF7 is regulated by auto-inhibition, as the full-length protein is inactive for microtubule binding in cells. We identify a segment, the inhibitory coiled coil (inhCC), that is required for auto-inhibition of KIF7, whereas the adjacent regulatory coiled coil (rCC) that contributes to auto-inhibition of the motile kinesin-4s KIF21A and KIF21B is not sufficient for KIF7 auto-inhibition. Disease-associated mutations in the inhCC relieve auto-inhibition and result in strong microtubule binding. Surprisingly, uninhibited KIF7 proteins did not bind preferentially to or track the plus ends of growing microtubules in cells, as suggested by previous in vitro work, but rather bound along cytosolic and axonemal microtubules. Localization to the tip of the primary cilium also required the inhCC, and could be increased by disease-associated mutations regardless of the auto-inhibition state of the protein. These findings suggest that loss of KIF7 auto-inhibition and/or altered cilium tip localization can contribute to the pathogenesis of human disease.
Publicações recentes
Midline defect with corpus callosum agenesis, vermian hypoplasia and median cleft lip palate.
Retinitis Pigmentosa Sine Pigmento in a Patient With a Heterozygous Mutation on the KIF7 Gene: A Case Report.
Intracranial arachnoid cysts.
Familial and syndromic forms of arachnoid cyst implicate genetic factors in disease pathogenesis.
Sequences in the stalk domain regulate auto-inhibition and ciliary tip localization of the immotile kinesin-4 KIF7.
📚 EuropePMC70 artigos no totalmostrando 16
Midline defect with corpus callosum agenesis, vermian hypoplasia and median cleft lip palate.
Case reports in perinatal medicineRetinitis Pigmentosa Sine Pigmento in a Patient With a Heterozygous Mutation on the KIF7 Gene: A Case Report.
CureusIntracranial arachnoid cysts.
Child's nervous system : ChNS : official journal of the International Society for Pediatric NeurosurgeryFamilial and syndromic forms of arachnoid cyst implicate genetic factors in disease pathogenesis.
Cerebral cortex (New York, N.Y. : 1991)Sequences in the stalk domain regulate auto-inhibition and ciliary tip localization of the immotile kinesin-4 KIF7.
Journal of cell scienceAcrocallosal Syndrome First Presenting with Acute Lymphoblastic Leukemia: A Rare Case Report.
Neurology IndiaOlfactory bulb and olfactory tract abnormalities in acrocallosal syndrome and Greig cephalopolysyndactyly syndrome.
Pediatric radiologyIntracranial cystic lesions and polydactyly associated with acrocallosal syndrome: Sonographic findings in two cases.
Journal of clinical ultrasound : JCUAltered GLI3 and FGF8 signaling underlies acrocallosal syndrome phenotypes in Kif7 depleted mice.
Human molecular geneticsAnaesthetising an infant with acrocallosal syndrome: An unusual case.
Indian journal of anaesthesiaClinical and experimental evidence suggest a link between KIF7 and C5orf42-related ciliopathies through Sonic Hedgehog signaling.
European journal of human genetics : EJHGAnterior Fontanelle Wormian Bone With Exomphalos Major and Dysmorphic Facial Features: A Previously Unseen Association?
The Journal of craniofacial surgeryNovel KIF7 Mutation in a Tunisian Boy with Acrocallosal Syndrome: Case Report and Review of the Literature.
Molecular syndromologyMOLAR TOOTH SIGN AND ACROCALLOSAL SYNDROME--A REPORT ON A POLISH FAMILY AND REVIEW OF KIF7 SYNDROMOLOGY.
Genetic counseling (Geneva, Switzerland)Novel KIF7 missense substitutions in two patients presenting with multiple malformations and features of acrocallosal syndrome.
American journal of medical genetics. Part AA novel KIF7 mutation in two affected siblings with acrocallosal syndrome.
Clinical dysmorphologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome acrocalosal.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome acrocalosal
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Midline defect with corpus callosum agenesis, vermian hypoplasia and median cleft lip palate.
- Retinitis Pigmentosa Sine Pigmento in a Patient With a Heterozygous Mutation on the KIF7 Gene: A Case Report.
- Intracranial arachnoid cysts.Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery· 2023· PMID 37466684mais citado
- Familial and syndromic forms of arachnoid cyst implicate genetic factors in disease pathogenesis.
- Sequences in the stalk domain regulate auto-inhibition and ciliary tip localization of the immotile kinesin-4 KIF7.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:36(Orphanet)
- OMIM OMIM:200990(OMIM)
- MONDO:0008708(MONDO)
- GARD:5721(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q4675304(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome acrocalosal
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata