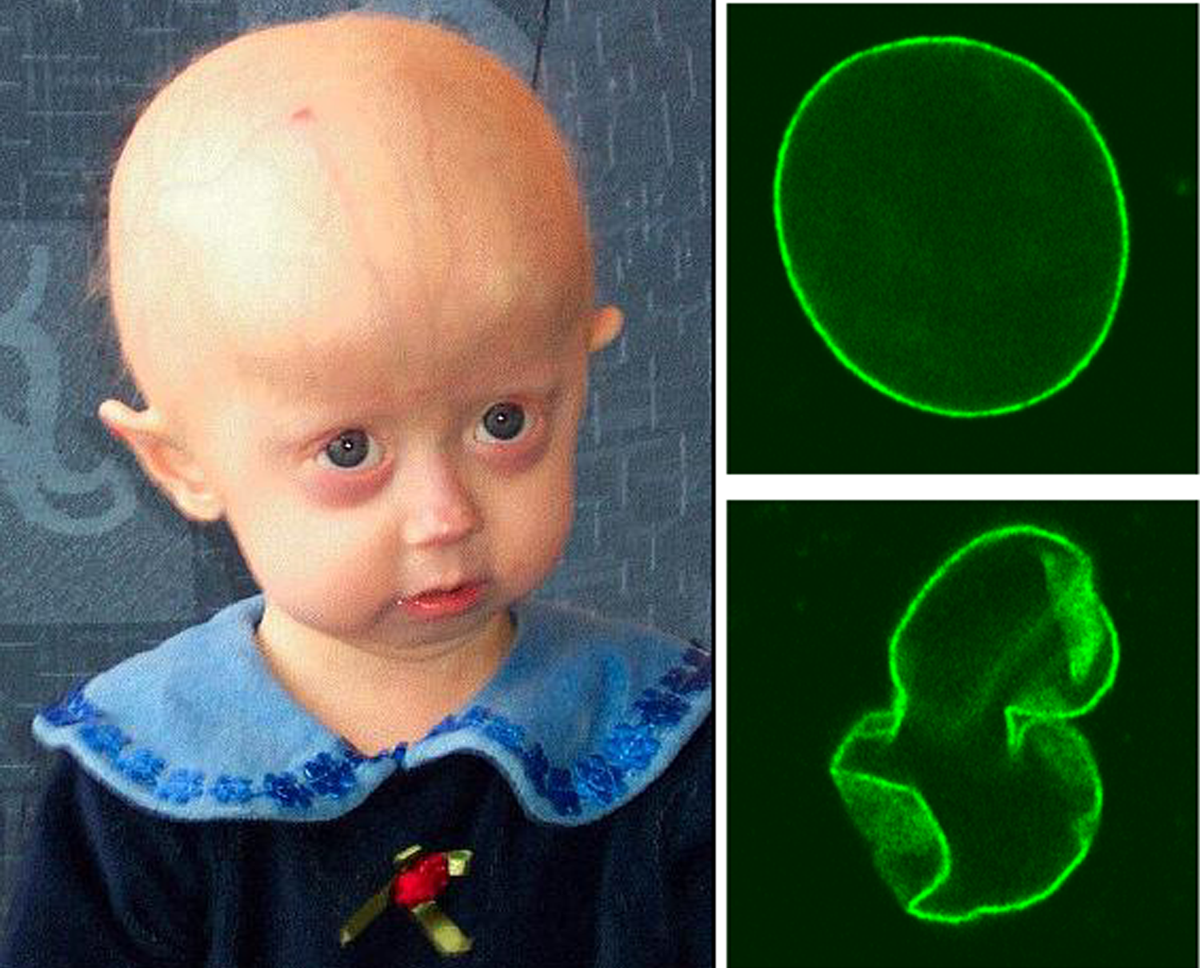
A Síndrome de Baller-Gerold é uma condição que se caracteriza pela combinação de uma fusão precoce dos ossos da parte da frente do crânio com anomalias na "linha" radial. Essas anomalias da "linha" radial (que envolvem os ossos e dedos do lado do polegar) podem incluir ter menos dedos que o normal, ausência total ou desenvolvimento incompleto do polegar, e ausência total ou desenvolvimento incompleto do osso rádio (um dos ossos do antebraço).
Introdução
O que você precisa saber de cara
A Síndrome de Baller-Gerold é uma condição que se caracteriza pela combinação de uma fusão precoce dos ossos da parte da frente do crânio com anomalias na "linha" radial. Essas anomalias da "linha" radial (que envolvem os ossos e dedos do lado do polegar) podem incluir ter menos dedos que o normal, ausência total ou desenvolvimento incompleto do polegar, e ausência total ou desenvolvimento incompleto do osso rádio (um dos ossos do antebraço).
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 41 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 113 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisAn ATP-dependent DNA helicase which unwinds dsDNA with a 3'-overhang in a 3'-5' direction (PubMed:28653661). Does not unwind more than 18 bp of dsDNA (PubMed:28653661). May modulate chromosome segregation. The N-terminal domain (residues 1-54) binds DNA Y-shaped DNA better than ss- or dsDNA (PubMed:22730300). The core helicase domain binds ssDNA (PubMed:22730300, PubMed:28653661)
CytoplasmNucleus
RAPADILINO syndrome
Disease characterized by radial and patellar aplasia or hypoplasia.
Variantes genéticas (ClinVar)
688 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 4.846 variantes classificadas pelo ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Baller-Gerold
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Molecular mechanism, diagnosis, and treatment of VACTERL association.
The VACTERL association is a non-random cluster of congenital malformations involving six distinct conditions: vertebral defects (V), anal atresia (A), cardiac defects (C), tracheoesophageal malformation (TE), renal defects (R), and limb anomalies (L), and is diagnosed when a fetus exhibits three or more of these. Its prevalence is approximately 0.47-0.58 per 10,000 live births. This paper examines the effect of disruptions in the Sonic Hedgehog and cilia-associated signaling pathways, genetically related developmental variations, and maternal environmental factors on the development of VACTERL. In the SHH signaling pathway, we focus on the effects of Sonic Hedgehog ligands, GLI transcription factors, and factors influencing GLI activity (RAC1 and ZIC3), as well as downstream targets (FOXF1 and HOXD13) and other genes and proteins involved in the regulation of SHH signaling (FGF8 and LPP), in the pathogenesis of VACTERL. In this context, ZIC3, which was shown to play a major role in VACTERL pathogenesis in large-scale resequencing, and TRAP1, which was associated with VACTERL pathogenesis in whole-exome resequencing, were highlighted. We also examine the cilia-associated signaling pathways, particularly the role of IFT172 and candidate ciliopathy genes. In addition, we describe the influence of TRAP1, COL11A2, SALL4, WBP11, Copy Number Variants, and maternal environmental factors on VACTERL. We also discuss current diagnostic, therapeutic, and prognostic approaches including prenatal and postnatal treatment options. Furthermore, we highlight the advantages of thoracoscopic surgery over traditional open-surgical treatment while discussing the differential diagnosis of VACTERL from other neonatal malformations with similar symptoms, such as Townes-Brocks syndrome, Baller-Gerold syndrome, and CHARGE syndrome.
Unilateral loss of recql4 function in Xenopus laevis tadpoles leads to ipsilateral ablation of the forelimb, hypoplastic Meckel's cartilage, and vascular defects.
RECQL4 encodes a RecQ helicase, one of a family of DNA unwinding enzymes with roles in DNA replication, double-strand break repair, and genomic stability. Pathogenic variants in RECQL4 are clinically associated with 3 rare autosomal recessive conditions: Rothmund-Thomson syndrome type II, Baller-Gerold syndrome, and RAPADILINO syndrome. These 3 syndromes show overlapping growth retardation, low bone density, and skeletal defects affecting the arms and hands. Here, we take advantage of the ability to generate one-sided CRISPR knockdowns of recql4 in Xenopus laevis tadpoles. Tadpoles develop normally until feeding starts, after which growth slows on the edited side, leading to a curved posture, smaller eyes (microphthalmia), and reduced head size (microcephaly). Forelimb buds fail to develop, leading to complete absence of the forelimb on the edited side. Additionally, Meckel's cartilage (lower jaw) ossification is absent or reduced and the hyoid cartilage is smaller, but this is not due to deficiencies in cranial neural crest migration on the edited side. Knockdown of recql4 also results in hypoplastic vasculature, with reduced branching from the aorta on the edited side. Taken together, our results clearly show the utility of unilateral CRISPR editing in Xenopus for understanding the specific phenotypic developmental effects of mutations affecting cell proliferation.
Minute amounts of helicase-deficient truncated RECQL4 are sufficient for DNA replication.
RECQL4 is a member of the RecQ family of helicases, playing essential roles in DNA replication and maintaining genome integrity. Mutations in RECQL4 are linked to severe human diseases, including Rothmund-Thomson Syndrome, RAPIDALINO Syndrome, and Baller-Gerold Syndrome. However, we still do not fully understand its functions and genetic interactions. The role of the ATP-dependent helicase activity in RECQL4 remains controversial. To understand RECQL4's functions further, we conducted a genome-wide forward genetic screen using murine models that closely mimic the RECQL4 mutations found in patients with Rothmund-Thomson syndrome. Our goal was to identify loss-of-function alleles that could rescue the proliferation and viability defects associated with RECQL4 mutation. From our screening we identified the loss of KLHDC3, a substrate-binding subunit of the Cullin-RING ligase (CRL) E3, as the most significant rescue allele. KLHDC3 facilitates the ubiquitin-mediated destruction of proteins with specific C-terminal degron motifs. Its loss normalized cell proliferation and DNA replication rates in cells with mutated RECQL4. Further analysis revealed that the loss of KLHDC3 led to the stabilization of minute levels of a truncated RECQL4 protein. This RECQL4 fragment contained a neo-degron sequence specific for KLHDC3, formed after Cre-mediated recombination of the Recql4 fl allele. Although this rescue mechanism does not apply to human RECQL4 mutations, it shows that very low chromatin-bound levels of a truncated RECQL4 protein-comprising only the N-terminal 480 amino acids, including its Sld2-like domain but lacking the ATP-dependent helicase domain and the entire C-terminal portion-are sufficient to support DNA replication in mammalian cells. These results demonstrate that the ATPase activity and helicase domain of RECQL4 are not essential for DNA replication in mammals. Furthermore, our findings suggest that there are unlikely to be monogenic loss-of-function alleles that can rescue RECQL4 mutations. This demonstrates that RECQL4 is an essential and non-redundant regulator of DNA replication and cell viability and that this activity does not require the ATP dependent helicase activity.
Severe Phenotype With RECQL4 Syndrome: A Report of Two Cases.
Baller-Gerold syndrome (BGS, OMIM: 218600), RAPADILINO syndrome (OMIM 266280), and Rothmund-Thomson syndrome (RTS, OMIM 266280), which are caused in some cases by RECQL4 pathogenic variants, show autosomal recessive inheritance. Some refer to them collectively as RECQL4 syndromes. Most cases have been reported during infancy and childhood periods. However, there have been no reports of phenotypes resulting in a lethal course in the perinatal period. We identified two fetuses with biallelic RECQL4 pathogenic variants during the perinatal period. The two fetuses with RECQL4 syndrome showed structural abnormalities, including severely hypoplastic forearms and lower legs. One fetus also had severe pulmonary hypoplasia. One case resulted in neonatal death because of respiratory failure, and the other was artificially terminated during pregnancy. The RECQL4 pathogenic variants were identified by exome sequencing followed by Sanger sequencing. The biallelic RECQL4 pathogenic variants can induce a lethal skeletal disorder.
Regression of Monosomy 7 Clone in Patient With RECQL4-Associated Syndrome.
Publicações recentes
Unilateral loss of recql4 function in Xenopus laevis tadpoles leads to ipsilateral ablation of the forelimb, hypoplastic Meckel's cartilage, and vascular defects.
Minute amounts of helicase-deficient truncated RECQL4 are sufficient for DNA replication.
Molecular mechanism, diagnosis, and treatment of VACTERL association.
Regression of Monosomy 7 Clone in Patient With RECQL4-Associated Syndrome.
Severe Phenotype With RECQL4 Syndrome: A Report of Two Cases.
📚 EuropePMC42 artigos no totalmostrando 17
Unilateral loss of recql4 function in Xenopus laevis tadpoles leads to ipsilateral ablation of the forelimb, hypoplastic Meckel's cartilage, and vascular defects.
G3 (Bethesda, Md.)Minute amounts of helicase-deficient truncated RECQL4 are sufficient for DNA replication.
bioRxiv : the preprint server for biologyMolecular mechanism, diagnosis, and treatment of VACTERL association.
Frontiers in pediatricsRegression of Monosomy 7 Clone in Patient With RECQL4-Associated Syndrome.
American journal of hematologySevere Phenotype With RECQL4 Syndrome: A Report of Two Cases.
American journal of medical genetics. Part AMolecular Mechanisms of the RECQ4 Pathogenic Mutations.
Frontiers in molecular biosciencesHuman RecQL4 as a Novel Molecular Target for Cancer Therapy.
Cytogenetic and genome researchHuman RecQ Helicases in DNA Double-Strand Break Repair.
Frontiers in cell and developmental biologyCongenital Diseases of DNA Replication: Clinical Phenotypes and Molecular Mechanisms.
International journal of molecular sciencesPathogenic variants in CDC45 on the remaining allele in patients with a chromosome 22q11.2 deletion result in a novel autosomal recessive condition.
Genetics in medicine : official journal of the American College of Medical GeneticsPhenotypic Overlap of Roberts and Baller-Gerold Syndromes in Two Patients With Craniosynostosis, Limb Reductions, and ESCO2 Mutations.
Frontiers in pediatricsRecQL4-Aurora B kinase axis is essential for cellular proliferation, cell cycle progression, and mitotic integrity.
OncogenesisHuman RecQL4 helicase plays multifaceted roles in the genomic stability of normal and cancer cells.
Cancer lettersNationwide survey of Baller‑Gerold syndrome in Japanese population.
Molecular medicine reportsIdentification of novel compound heterozygous RECQL4 mutations and prenatal diagnosis of Baller-Gerold syndrome: a case report.
Genetics and molecular research : GMRThe DNA helicase recql4 is required for normal osteoblast expansion and osteosarcoma formation.
PLoS geneticsRECQL4 Regulates p53 Function In Vivo During Skeletogenesis.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Baller-Gerold.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Baller-Gerold
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Molecular mechanism, diagnosis, and treatment of VACTERL association.
- Unilateral loss of recql4 function in Xenopus laevis tadpoles leads to ipsilateral ablation of the forelimb, hypoplastic Meckel's cartilage, and vascular defects.
- Minute amounts of helicase-deficient truncated RECQL4 are sufficient for DNA replication.
- Severe Phenotype With RECQL4 Syndrome: A Report of Two Cases.
- Regression of Monosomy 7 Clone in Patient With RECQL4-Associated Syndrome.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1225(Orphanet)
- OMIM OMIM:218600(OMIM)
- MONDO:0009039(MONDO)
- GARD:1602(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q3508616(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome Baller-Gerold
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata