Anomalia cromossômica caracterizada por características dismórficas faciais distintas, hipotonia, atraso no desenvolvimento, deficiência intelectual, convulsões, defeitos cardíacos, deficiência auditiva e deficiência de crescimento de início pré-natal.
Introdução
O que você precisa saber de cara
Anomalia cromossômica caracterizada por características dismórficas faciais distintas, hipotonia, atraso no desenvolvimento, deficiência intelectual, convulsões, defeitos cardíacos, deficiência auditiva e deficiência de crescimento de início pré-natal.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 51 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 156 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
13 genes identificados com associação a esta condição. Padrão de herança: Multigenic/multifactorial, Not applicable.
F-actin cross-linking protein (PubMed:30990684). Stabilizes actin and acts as a negative regulator of primary cilium formation (PubMed:32496561). Positively regulates the phosphorylation of both myosin II and protein phosphatase 1 regulatory subunit PPP1R12A/MYPT1 and promotes the assembly of myosin II stacks within actin stress fibers (PubMed:38832964). Inhibits the phosphorylation of myosin light chain MYL9 by DAPK3 and suppresses the constriction velocity of the contractile ring during cytoki
Cytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, cilium basal bodyMidbodyChromosome, centromereCytoplasm, cytoskeleton, spindleCytoplasm, cytoskeleton, stress fiberNucleusCell projection, dendritePerikaryonCell junction, tight junction
Transcriptional activator (PubMed:23639441, PubMed:27693370). Involved in vascular assembly and morphogenesis through direct transcriptional regulation of EGFL7 (PubMed:23639441)
Nucleus
May play a role in terminal differentiation of skeletal muscle cells but not in the determination of cells to the myogenic lineage. Functions as a repressor of TGF-beta signaling
Nucleus
Shprintzen-Goldberg craniosynostosis syndrome
A very rare syndrome characterized by a marfanoid habitus, craniosynostosis, characteristic dysmorphic facial features, skeletal and cardiovascular abnormalities, intellectual disability, developmental delay and learning disabilities.
Calcium- and diacylglycerol-independent serine/threonine-protein kinase that functions in phosphatidylinositol 3-kinase (PI3K) pathway and mitogen-activated protein (MAP) kinase cascade, and is involved in NF-kappa-B activation, mitogenic signaling, cell proliferation, cell polarity, inflammatory response and maintenance of long-term potentiation (LTP). Upon lipopolysaccharide (LPS) treatment in macrophages, or following mitogenic stimuli, functions downstream of PI3K to activate MAP2K1/MEK1-MAP
CytoplasmEndosomeCell junctionMembrane
May serve as a nuclear matrix platform that organizes and integrates transcriptional responses. In osteoblasts, supports transcription activation: synergizes with RUNX2 to enhance FGFR2-mediated activation of the osteocalcin FGF-responsive element (OCFRE) (By similarity). Has also been shown to be an essential corepressor protein, which probably regulates different key pathways such as the Notch pathway. Negative regulator of the Notch pathway via its interaction with RBPSUH, which prevents the
Nucleus
Radio-Tartaglia syndrome
An autosomal dominant neurodevelopmental disorder characterized by global developmental delay, hypotonia, mild motor difficulties, impaired intellectual development, speech delay, craniofacial dysmorphism, and variable behavioral abnormalities.
Transcription regulator that acts both as a histone methyltransferase or chromatin adapter, depending on the context (PubMed:12816872). In the cytoplasm, acts as a histone methyltransferase, which catalyzes monomethylation of 'Lys-9' of free histone H3 (H3K9me1) during translation (By similarity). Monomethylated histone H3 is then transported to the nucleus and incorporated into nucleosomes where SUV39H methyltransferases (SUV39H1 and SUV39H2) use it as a substrate to catalyze histone H3 'Lys-9'
NucleusChromosomeCytoplasm
Left ventricular non-compaction 8
A form of left ventricular non-compaction, a cardiomyopathy due to myocardial morphogenesis arrest and characterized by a hypertrophic left ventricle, a severely thickened 2-layered myocardium, numerous prominent trabeculations, deep intertrabecular recesses, and poor systolic function. Clinical manifestations are variable. Some affected individuals experience no symptoms at all, others develop heart failure. In some cases, left ventricular non-compaction is associated with other congenital heart anomalies. LVNC8 is an autosomal dominant condition.
Integral component of basement membranes. Component of the glomerular basement membrane (GBM), responsible for the fixed negative electrostatic membrane charge, and which provides a barrier which is both size- and charge-selective. It serves as an attachment substrate for cells. Plays essential roles in vascularization. Critical for normal heart development and for regulating the vascular response to injury. Also required for avascular cartilage development (PubMed:12435733, PubMed:15591058, Pub
Secreted, extracellular space, extracellular matrix, basement membraneSecreted
Schwartz-Jampel syndrome
Rare autosomal recessive disorder characterized by permanent myotonia (prolonged failure of muscle relaxation) and skeletal dysplasia, resulting in reduced stature, kyphoscoliosis, bowing of the diaphyses and irregular epiphyses.
Regulatory subunit of the voltage-gated potassium (Kv) Shaker channels composed of pore-forming and potassium-conducting alpha subunits and of regulatory beta subunits (PubMed:11825900, PubMed:7649300). The beta-2/KCNAB2 cytoplasmic subunit promotes potassium channel closure via a mechanism that does not involve physical obstruction of the channel pore (PubMed:11825900, PubMed:7649300). Promotes the inactivation of Kv1.4/KCNA4 and Kv1.5/KCNA5 alpha subunit-containing channels (PubMed:11825900, P
CytoplasmMembraneCell membraneCell projection, axonSynapse, synaptosomeCytoplasm, cytoskeleton
Protease. May regulate the surface expression of some potassium channels by retaining them in the endoplasmic reticulum (By similarity)
Endoplasmic reticulum membraneMembrane
Mediates effects on cell migration and adhesion through its different partners. During development plays a role in blood and lymphatic vessels separation by binding CLEC1B, triggering CLEC1B activation in platelets and leading to platelet activation and/or aggregation (PubMed:14522983, PubMed:15231832, PubMed:17222411, PubMed:17616532, PubMed:18215137). Interaction with CD9, on the contrary, attenuates platelet aggregation induced by PDPN (PubMed:18541721). Through MSN or EZR interaction promote
MembraneCell projection, lamellipodium membraneCell projection, filopodium membraneCell projection, microvillus membraneCell projection, ruffle membraneMembrane raftApical cell membraneBasolateral cell membraneCell projection, invadopodiumCytoplasm, cytosol
Plays a role as a transcriptional repressor during development. May play a role in the control of cell survival. Overexpression of RERE recruits BAX to the nucleus particularly to POD and triggers caspase-3 activation, leading to cell death
Nucleus
Ubiquitin-protein ligase that probably functions as an E3 ligase in conjunction with specific E1 and E2 ligases (By similarity). May also function as an E4 ligase mediating the assembly of polyubiquitin chains on substrates ubiquitinated by another E3 ubiquitin ligase (By similarity). May regulate myosin assembly in striated muscles together with STUB1 and VCP/p97 by targeting myosin chaperone UNC45B for proteasomal degradation (PubMed:17369820)
CytoplasmNucleus
Delta subunit of the heteropentameric ligand-gated chloride channel gated by gamma-aminobutyric acid (GABA), a major inhibitory neurotransmitter in the brain (PubMed:35355020). GABA-gated chloride channels, also named GABA(A) receptors (GABAAR), consist of five subunits arranged around a central pore and contain GABA active binding site(s) located at the alpha and beta subunit interface(s) (PubMed:35355020). When activated by GABA, GABAARs selectively allow the flow of chloride anions across the
Cell membrane
Generalized epilepsy with febrile seizures plus 5
A rare autosomal dominant, familial condition with incomplete penetrance and large intrafamilial variability. Patients display febrile seizures persisting sometimes beyond the age of 6 years and/or a variety of afebrile seizure types. This disease combines febrile seizures, generalized seizures often precipitated by fever at age 6 years or more, and partial seizures, with a variable degree of severity.
Variantes genéticas (ClinVar)
453 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 15 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
35 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de deleção 1p36
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
2 ensaios clínicos encontrados, 1 ativos.
Publicações mais relevantes
First Detection of 1p36 Deletion by Whole-Exome Sequencing in a Tunisian Patient.
We reported a rare case of 1p36 deletion syndrome diagnosed using whole-exome sequencing (WES) in a Tunisian neonate, and to highlight the utility of WES in detecting structural variants, particularly in resource-limited settings. Clinical and genetic investigations were conducted on a female neonate presenting with a severe polymalformative syndrome. WES was performed to detect potential genetic abnormalities, followed by validation through fluorescence in situ hybridization (FISH). Variant annotation and classification were done in accordance with ACMG guidelines. WES identified a heterozygous interstitial deletion in the 1p36 region, spanning 11.64 Mb and affecting 155 coding genes, including key genes such as MMP23B, GABRD, SKI, PRDM16, KCNAB2, RERE, UBE4B, and CASZ1. The deletion was classified as pathogenic, and FISH analysis confirmed its presence. Clinically, the patient exhibited intrauterine growth restriction, neonatal epilepsy, craniofacial dysmorphia, congenital heart defect, and agenesis of the corpus callosum. This is the first reported case in Tunisia of a 1p36 deletion identified via short-read WES. The findings support the expanding role of WES in structural variant detection and underscore its diagnostic value, especially in settings with limited access to chromosomal microarray or genome sequencing technologies.
Two Cases of SPEN Haploinsufficiency Presenting with Dystonia: Expanding the Genotype and Phenotype.
Dental Anomalies in 1p36 Deletion Syndrome: A Case Report.
This report describes a nine-year, five-month-old Saudi girl with 1p36 deletion syndrome (1p36DS) referred for dental evaluation due to esthetic concerns. Clinical and radiographic assessment revealed multiple carious lesions, poor oral hygiene, dens invaginatus in maxillary incisors, infraocclusion of a primary molar, and, notably, agenesis of all primary and permanent canines except for one retained mandibular primary canine. Additionally, two supernumerary maxillary incisors (mesiodens) were identified, causing severe rotation of adjacent teeth, a combination of dental anomalies not previously documented in 1p36DS. A comprehensive treatment plan was executed, encompassing preventive measures, restorative care, surgical extraction of mesiodens, and interceptive orthodontic alignment using a 2×4 fixed appliance and transpalatal arch. These interventions led to improved esthetics, dental function, and psychosocial confidence. The management of 1p36DS patients requires a multidisciplinary approach due to the conjunction of craniofacial and systemic anomalies. Pediatric dentists play a crucial role in early detection, preventive care, and timely interventions, all of which contribute to enhanced function and quality of life. Routine panoramic imaging and vigilance for atypical dental patterns are critical for guiding genetic referrals and comprehensive care. This case broadens the known phenotypic spectrum of 1p36DS by documenting a novel dental presentation involving concurrent agenesis of both primary and permanent canines with supernumerary maxillary incisors. Early recognition of such anomalies enables timely diagnosis, personalized management, and multidisciplinary collaboration, emphasizing the vital role of pediatric dentists in the holistic care of patients with rare chromosomal disorders.
Prenatally diagnosed chromosome 1p36 deletions: a retrospective case series, literature review, and genotype-phenotype correlations.
Chromosome 1p36 deletion syndrome is the most common terminal autosomal deletion disorder. While postnatal features are well-defined, prenatal characterization remains less comprehensive. This study aims to delineate the prenatal sonographic and genetic spectrum of this syndrome and explore genotype-phenotype correlations. A retrospective analysis was conducted on 21 consecutive prenatally diagnosed cases of 1p36 deletion (2017–2025) from a single tertiary center. Data on maternal demographics, ultrasound findings, genetic results, inheritance patterns, and pregnancy outcomes were collected. A pooled analysis included 10 isolated cases (with complete ultrasound data) from our cohort and 33 previously reported cases. The cohort demonstrated a strong female predominance (71.4%, 15/21). Fifteen (71.4%) involved isolated 1p36 deletions, while six (28.6%) exhibited additional chromosomal aberrations. Pooled analysis of 43 prenatally diagnosed isolated cases (10 current and 33 published) revealed brain anomalies to be the most common prenatal feature (58.1%, 25/43), with ventriculomegaly (37.2%, 16/43), corpus callosum hypoplasia (9.3%, 4/43), and choroid plexus cysts (9.3%, 4/43) being the most frequent subtypes. Cardiac defects were observed in 39.5% (17/43) of cases, predominantly atrial/ventricular septal defects (ASD/VSD, 23.3%, 10/43) and Ebstein anomaly (11.6%, 5/43). Other frequently observed features included increased nuchal translucency (16.3%, 7/43) and single umbilical artery (14%, 6/43). Parental genetic analysis demonstrated de novo deletions in 80.0% (12/15) of cases, while 20.0% (3/15) were inherited from phenotypically normal parents. Congenital brain anomalies (particularly ventriculomegaly) and cardiac defects are hallmark prenatal features of isolated 1p36 deletion syndrome. Our findings underscore the role of key genes (RERE, SPEN, PRDM16, MMP23B) in shaping the phenotype and highlight the importance of parental genetic testing in counseling, given the potential for inherited variants with variable expressivity.
Ultrasound Phenotype, Genetic Analysis, and Pregnancy Outcomes of Fetuses With 1p36 Deletion Syndrome.
The intrauterine ultrasound phenotype, genotype, pregnancy outcome, and neonatal prognosis of fetuses with 1p36 deletion syndrome were retrospectively analyzed, as previous reports are limited. Pregnant women (25,000) who underwent interventional prenatal diagnosis between December 2016 and March 2024 were selected. Fetal villus tissue, amniotic fluid, or umbilical cord blood were extracted for single nucleotide polymorphism array (SNP-array) detection under ultrasound guidance. Thirteen fetuses had 1p36 deletions involving fragments that were 0.46-22.5 Mb. Six and seven fetuses had large and small copy number variation (CNV) fragment deletions in the 1p36 region, respectively. Two fetuses had normal ultrasound phenotypes, three underwent early spontaneous abortion, one had isolated ventricular septal defect, one had isolated mild ventriculomegaly, two had mild ventriculomegaly associated with increased renal echogenicity, one had mild ventriculomegaly associated with ventricular septal defect, one had severe ventriculomegaly associated with ventricular septal defect and fetal growth restriction, one had tricuspid valve dysplasia, and one had nasal bone dysplasia. Three 1p36 deletions were de novo, and one was paternally inherited. There were three cases of early spontaneous abortion, seven terminations, and three routine postnatal follow-ups. High-resolution SNP-arrays are suitable for the prenatal diagnosis of 1p36 deletion syndrome.
Publicações recentes
An enhanced transformer model for detecting 1p36 deletion syndrome.
First Detection of 1p36 Deletion by Whole-Exome Sequencing in a Tunisian Patient.
🥇 Revisão sistemáticaTwo Cases of SPEN Haploinsufficiency Presenting with Dystonia: Expanding the Genotype and Phenotype.
Dental Anomalies in 1p36 Deletion Syndrome: A Case Report.
Prenatally diagnosed chromosome 1p36 deletions: a retrospective case series, literature review, and genotype-phenotype correlations.
📚 EuropePMC71 artigos no totalmostrando 63
First Detection of 1p36 Deletion by Whole-Exome Sequencing in a Tunisian Patient.
Birth defects researchTwo Cases of SPEN Haploinsufficiency Presenting with Dystonia: Expanding the Genotype and Phenotype.
Movement disorders clinical practiceDental Anomalies in 1p36 Deletion Syndrome: A Case Report.
CureusPrenatally diagnosed chromosome 1p36 deletions: a retrospective case series, literature review, and genotype-phenotype correlations.
BMC medical genomicsUltrasound Phenotype, Genetic Analysis, and Pregnancy Outcomes of Fetuses With 1p36 Deletion Syndrome.
Molecular genetics & genomic medicineAnesthesia management for dental procedures in a patient with 1p36 deletion syndrome: a case report.
Journal of dental anesthesia and pain medicineFirst-trimester application of expanded non-invasive prenatal testing in the genetic investigation of fetal 1p36 deletion syndrome associated with a familial unbalanced reciprocal translocation of 46,XX,der(1)t(1;2) (p36.2;q37.3)dmat.
Taiwanese journal of obstetrics & gynecologyDNA methylation dysregulation patterns in the 1p36 region instability.
Journal of applied geneticsBreaking new ground: Exploring de novo chromosomal rearrangements in 1p36 microdeletion.
International journal of health sciences[Prenatal diagnosis of a fetus with 1p36 deletion syndrome and 3p26.3p25.2 duplication].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsPRDM16 co-operates with LHX2 to shape the human brain.
Oxford open neuroscience[Analysis of genetic etiology in a patient with 1p36 deletion syndrome in conjunct with Snijders Blok-Campeau syndrome].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsGenetic Analysis of 1p36 Deletions for Six Aborted Fetuses.
Alternative therapies in health and medicineCongenital diaphragmatic hernia in patient with 1p36 deletion.
Clinical case reportsNoninsulinoma Pancreatogenous Hypoglycemia Syndrome in a Patient With 1p36 Deletion Syndrome.
JCEM case reportsDeletion in 1p36.33-p36.32 is associated with pancytopenia: a case report.
BMC medical genomicsA Rare Case of an Infant With 1p36 Deletion Syndrome Presenting With Systolic Heart Failure Secondary to Severe Dilated Cardiomyopathy.
CureusPRDM16 Deletion Is Associated With Sex-dependent Cardiomyopathy and Cardiac Mortality: A Translational, Multi-Institutional Cohort Study.
Circulation. Genomic and precision medicine1p36 deletion syndrome: Review and mapping with further characterization of the phenotype, a new cohort of 86 patients.
American journal of medical genetics. Part AOutcome of Vertical Expandable Prosthetic Titanium Rib (VEPTR) Instrumentation in Scoliosis Associated With 1p36 Deletion Syndrome: A Case Report.
CureusAbatacept as an alternative therapy for the treatment of drug-intolerant lupus nephritis: A case of underlying monosomy 1p36 deletion syndrome.
Clinical nephrologyPrenatal diagnosis of pure 1p36 terminal deletion by chromosome microarry analysis - clinical report of 3 new cases and review of the literature.
Ginekologia polskaMonosomy 1p36: Report of a cohort of 13 Asian Indian patients.
American journal of medical genetics. Part APsychiatric Comorbidities in 1p36 Deletion Syndrome and Their Treatment-A Case Report.
International journal of environmental research and public health1p36 Deletion Syndrome and the Aorta: A Report of Three New Patients and a Literature Review.
Journal of cardiovascular development and diseaseNovel Point Mutation of EBSS Gene Coexisted with 1p36 Deletion.
Annals of dermatologyNoncongenital juvenile-onset bilateral lamellar cataract in 1p36 deletion syndrome.
Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and StrabismusThe Neuropathology of 1p36 Deletion Syndrome: An Autopsy Case Series.
Journal of neuropathology and experimental neurology1p36 Deletion Syndrome and Left Ventricular Non-compaction Cardiomyopathy-Two Cases Report.
Frontiers in pediatricsRERE deficiency contributes to the development of orofacial clefts in humans and mice.
Human molecular genetics1p36 deletion syndrome: first case report in Morocco detected by fluorescence in situ hybridization.
The Pan African medical journalSPEN haploinsufficiency causes a neurodevelopmental disorder overlapping proximal 1p36 deletion syndrome with an episignature of X chromosomes in females.
American journal of human geneticsA rare cardiac phenotype of dextrocardia observed in a fetus with 1p36 deletion syndrome and a balanced translocation: a prenatal case report.
Molecular cytogeneticsNon-invasive prenatal testing (NIPT) by low coverage genomic sequencing: Detection limits of screened chromosomal microdeletions.
PloS oneDental anomalies as a possible clue of 1p36 deletion syndrome due to germline mosaicism: a case report.
BMC pediatricsA new 1p36.13-1p36.12 microdeletion syndrome characterized by learning disability, behavioral abnormalities, and ptosis.
Clinical geneticsLarge 1p36 Deletions Affecting Arid1a Locus Facilitate Mycn-Driven Oncogenesis in Neuroblastoma.
Cell reportsPrenatal detection of 1p36 deletion syndrome: ultrasound findings and microarray testing results.
The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal ObstetriciansPerinatal distress in 1p36 deletion syndrome can mimic hypoxic ischemic encephalopathy.
American journal of medical genetics. Part APrenatal findings in 1p36 deletion syndrome: New cases and a literature review.
Prenatal diagnosisRer1-mediated quality control system is required for neural stem cell maintenance during cerebral cortex development.
PLoS geneticsElectroclinical features of epilepsy monosomy 1p36 syndrome and their implications.
Acta neurologica ScandinavicaIdentification of a New Candidate Locus for Ebstein Anomaly in 1p36.2.
Molecular syndromologyCutis laxa in a patient with 1p36 deletion syndrome.
The Journal of dermatology1p deletion syndrome: A prenatal diagnosis characterized by an abnormal 1st trimester combined screening test, yet a normal NIPT result.
Taiwanese journal of obstetrics & gynecologyGenotype-phenotype correlations in individuals with pathogenic RERE variants.
Human mutationElectroclinical features of epilepsy associated with 1p36 deletion syndrome: A review.
Epilepsy research[Prenatal diagnosis of two fetuses with chromosome 1p36 deletion syndrome].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsEarly transition from insulin to sulfonylureas in neonatal diabetes and follow-up: Experience from China.
Pediatric diabetes1p36 deletion syndrome confirmed by fluorescence in situ hybridization and array-comparative genomic hybridization analysis.
Korean journal of pediatrics[An updated review of 1p36 deletion (monosomy) syndrome].
Revista chilena de pediatriaAbdominal paraganglioma in a young woman with 1p36 deletion syndrome.
American journal of medical genetics. Part AGenomic findings in patients with clinical suspicion of 22q11.2 deletion syndrome.
Journal of applied geneticsThe TNF Receptor Superfamily in Co-stimulating and Co-inhibitory Responses.
ImmunityIs 1p36 deletion associated with anterior body wall defects?
American journal of medical genetics. Part ADe Novo Mutations of RERE Cause a Genetic Syndrome with Features that Overlap Those Associated with Proximal 1p36 Deletions.
American journal of human geneticsCharacterization of a variant of t(14;18) negative nodal diffuse follicular lymphoma with CD23 expression, 1p36/TNFRSF14 abnormalities, and STAT6 mutations.
Modern pathology : an official journal of the United States and Canadian Academy of Pathology, IncMini-Review: Monosomy 1p36 syndrome: reviewing the correlation between deletion sizes and phenotypes.
Genetics and molecular research : GMRIdentification of 1p36 deletion syndrome in patients with facial dysmorphism and developmental delay.
Korean journal of pediatrics1p36 deletion syndrome: an update.
The application of clinical geneticsLeft Ventricular Non-compaction: Is It Genetic?
Pediatric cardiologyAre Angelman and Prader-Willi syndromes more similar than we thought? Food-related behavior problems in Angelman, Cornelia de Lange, fragile X, Prader-Willi and 1p36 deletion syndromes.
American journal of medical genetics. Part ALeft ventricular noncompaction cardiomyopathy: adult association with 1p36 deletion syndrome.
Methodist DeBakey cardiovascular journalAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de deleção 1p36.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de deleção 1p36
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- First Detection of 1p36 Deletion by Whole-Exome Sequencing in a Tunisian Patient.
- Two Cases of SPEN Haploinsufficiency Presenting with Dystonia: Expanding the Genotype and Phenotype.
- Dental Anomalies in 1p36 Deletion Syndrome: A Case Report.
- Prenatally diagnosed chromosome 1p36 deletions: a retrospective case series, literature review, and genotype-phenotype correlations.
- Ultrasound Phenotype, Genetic Analysis, and Pregnancy Outcomes of Fetuses With 1p36 Deletion Syndrome.
- An enhanced transformer model for detecting 1p36 deletion syndrome.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1606(Orphanet)
- OMIM OMIM:607872(OMIM)
- MONDO:0011929(MONDO)
- GARD:6082(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q3297103(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
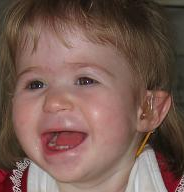
Síndrome de deleção 1p36
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov