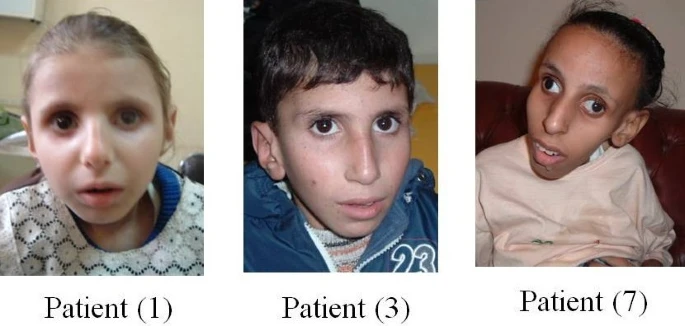
É uma síndrome rara, de origem genética, que causa múltiplas alterações no corpo desde o nascimento e características físicas distintas. Ela se manifesta com dificuldade de coordenação dos movimentos (ataxia), sensibilidade à luz (principalmente no rosto e tronco), baixa estatura e deficiência intelectual. Outras características que podem ser observadas são: dedos curvos, uma única linha na palma da mão, céu da boca (palato) muito alto ou arqueado, panturrilhas com aspecto aumentado (mas que não são de fato mais musculosas) e problemas na válvula aórtica do coração. Não houve mais descrições desta síndrome na literatura médica desde 1983.
Introdução
O que você precisa saber de cara
É uma síndrome rara, de origem genética, que causa múltiplas alterações no corpo desde o nascimento e características físicas distintas. Ela se manifesta com dificuldade de coordenação dos movimentos (ataxia), sensibilidade à luz (principalmente no rosto e tronco), baixa estatura e deficiência intelectual. Outras características que podem ser observadas são: dedos curvos, uma única linha na palma da mão, céu da boca (palato) muito alto ou arqueado, panturrilhas com aspecto aumentado (mas que não são de fato mais musculosas) e problemas na válvula aórtica do coração. Não houve mais descrições desta síndrome na literatura médica desde 1983.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 7 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 16 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de ataxia-fotossensibilidade-baixa estatura
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Phosphoproteomics elucidates the functional impact of the PTPN11 p.Asn308Ser variant in a Noonan syndrome pedigree.
Noonan syndrome (NS) is a common autosomal dominant disorder with considerable clinical heterogeneity. Mutations in the PTPN11 gene, encoding the SHP2 protein, constitute the most prevalent genetic cause of NS. Genetic sequencing of a pedigree exhibiting typical facial dysmorphism and short stature identified the same heterozygous PTPN11 variant (NM_001330437.2: c.923 A > G, p.Asn308Ser) in all seven affected individuals, co-segregating with the phenotype. Using a multi-level approach that integrated experimental structural analysis, molecular dynamics simulations, protein-protein interaction network analysis, quantitative phosphoproteomics, and functional validation in cellular models, we systematically elucidated the pathogenic mechanism of the p.Asn308Ser mutation. This revealed that the mutation disrupts critical hydrogen bonds and remodels the interaction network. This change enhances conformational heterogeneity and shifts the protein into an activated "open" state. Consequently, the mutation strengthens interactions with hub proteins, such as GRB2 and SRC, resulting in sustained RAS/MAPK activation. Phosphoproteomic analysis showed that the mutation induces extensive phosphorylation events, with differentially phosphorylated proteins significantly enriched in the nucleus, particularly in pathways related to chromatin organization and ATP-dependent chromatin remodeling. Further functional validation indicated that aberrantly activated ERK may phosphorylate chromatin remodeling complexes such as SWI/SNF, thereby directly connecting cytoplasmic signaling to aberrant epigenetic regulation in the nucleus. This study delineates the complete pathogenic axis of the PTPN11 p.Asn308Ser mutation, spanning atomic conformational changes and sustained signaling activation to aberrant nuclear chromatin remodeling. These findings extend the understanding of NS pathophysiology to the epigenetic level and provide a theoretical foundation for future interventions targeting both signaling pathways and chromatin states.
Case Report: Compound heterozygous mutations in the IDUA gene causing mucopolysaccharidosis type I with uterine developmental abnormality.
Mucopolysaccharidosis (MPS) represents a group of rare inherited metabolic disorders characterized by abnormal accumulation of glycosaminoglycans (GAGs) due to deficiencies of lysosomal enzymes. Mucopolysaccharidosis type I (MPS I) is caused by biallelic pathogenic variants in the IDUA gene and is inherited in an autosomal recessive pattern. The IDUA gene is located on chromosome 4p16.3 and encodes the lysosomal enzyme α-L-iduronidase, which plays a critical role in the degradation of GAGs, particularly dermatan sulfate and heparan sulfate. Reduced or absent IDUA enzymatic activity leads to the progressive accumulation of undegraded substrates within lysosomes, resulting in multisystem organ involvement. Based on clinical severity, MPS I is traditionally classified into three phenotypic subtypes: the severe form (Hurler syndrome), the intermediate form (Hurler-Scheie syndrome), and the attenuated form (Scheie syndrome, MPS I-S). This report describes a 13-year-old female patient in whom compound heterozygous pathogenic variants in the IDUA gene were identified by genetic testing, and whose clinical manifestations were consistent with the MPS I-S. In addition to typical skeletal and joint abnormalities, the patient also presented with uterine developmental abnormality. Currently, there is no definitive evidence supporting a direct causal relationship between MPS I and uterine developmental abnormalities; however, this case suggests a potential association between MPS I and reproductive system developmental abnormalities. This case may help further expand the phenotypic spectrum of MPS I and enhance clinical awareness of its multisystem involvement.
A Diagnostic Dilemma: Concurrent Diagnosis of Cystic Fibrosis and Definitive Kabuki Syndrome Type 1.
The article presents a clinical case involving a patient with presumptive coexistence of two hereditary disorders, confirmed by molecular genetic analyses. Clinical evaluation of the proband, a 9-year-old girl, revealed features characteristic of Kabuki syndrome, including a typical "Kabuki makeup" facial phenotype, short stature, intracranial hypertension, and diffuse muscular hypotonia. Additional clinical findings included chronic right-sided otitis media, conjunctivitis, recurrent pneumonia, bilateral conductive hearing loss, astigmatism, and primary adenitis. Clinical assessment and molecular genetic testing were performed. High-throughput sequencing identified a previously reported pathogenic heterozygous variant in the KMT2D gene, NM_003482.4:c.15142C>T p.Arg5048Cys, and two known heterozygous variants in the CFTR gene: NM_000492.4:c.1521_1523delCTT p.Phe508del and c.3454G>C p.Asp1152His, classified as pathogenic and of variable clinical significance, respectively. Segregation analysis demonstrated that the KMT2D variant most likely arose in the proband de novo, whereas the CFTR variants were inherited from each of the parents. Notably, the proband's clinically unaffected elder sister carried the same CFTR genotype. Based on the clinical presentation and molecular genetic findings, the diagnosis of Kabuki syndrome type 1 was conclusively established in the patient. Functional assessment of CFTR demonstrated its preserved function, which did not support a diagnosis of CF or CFTR-related disorders. PTDSS1-related Lenz-Majewski hyperostotic dysplasia (LMHD) is characterized by cutis laxa and progressive bone sclerosis, primarily affecting the skull and long bones. Individuals with classic PTDSS1-related LMHD typically presents in infancy with cutis laxa, prominent cutaneous veins, characteristic craniofacial features (disproportionately large head, broad forehead, delayed closure of the fontanelles, hypertelorism, large floppy ears, nasal obstruction / choanal atresia, macrostomia, thin vermilion of the lips, dental enamel hypoplasia, and prognathism or retrognathia), brachydactyly and syndactyly of the digits, early-onset osteosclerosis (involving the skull, spine, diaphyses of the long bones, clavicles, and ribs), severe growth deficiency, and significant developmental delays. Additional features can include genitourinary anomalies in males, inguinal hernia, ophthalmologic manifestations, hearing loss, and hydrocephalus. Attenuated PTDSS1-related LMHD is characterized by minimal or mild cutis laxa, slower progression of hyperostosis, preserved or mildly affected development, and normal stature. The diagnosis of PTDSS1-related LMHD is established in a proband with characteristic clinical and imaging findings and a heterozygous pathogenic gain-of-function variant in PTDSS1 identified by molecular genetic testing. Treatment of manifestations: Treatment of skeletal manifestations per orthopedist; consider physical therapy, occupational therapy, and assisted devices for mobility; decompression of cervical spine stenosis as needed; treatment of hydrocephalus as needed per neurosurgeon; treatment of respiratory difficulty and obstructive sleep apnea per otolaryngologist and/or pulmonologist; careful airway evaluation prior to surgical procedures; supportive therapies for those with developmental delays; individualized education plan for learning disorders and school performance issues; treatment of dental enamel hypoplasia per dentist; standard treatments for genitourinary anomalies, delayed puberty, inguinal hernia, vision issues, and hearing loss; consider dermatology referral for cosmetic concerns due to cutis laxa. Surveillance: Growth assessment and orthopedic evaluation annually or as determined by the orthopedist to monitor joint and skeletal manifestations; brain and spine MRI as needed; assess for manifestations of sleep apnea at each visit; polysomnography as needed; dental evaluation with frequency per dentist; ophthalmology evaluation with frequency per ophthalmologist; audiology evaluation as needed; monitor developmental progress, educational needs, and family needs at each visit. Agents/circumstances to avoid: Activities/procedures that involve extreme neck extension and flexion in individuals with craniovertebral junction stenosis. PTDSS1-related LMHD is an autosomal dominant disorder. All probands reported to date with PTDSS1-related LMHD whose parents have undergone molecular genetic testing have had the disorder as the result of a de novo PTDSS1 pathogenic variant. Risk to the parents of the proband of having another affected pregnancy is presumed to be low as the proband most likely has a de novo PTDSS1 pathogenic variant. There is, however, a recurrence risk (~1%) to sibs based on the possibility of parental gonadal mosaicism. Given this risk, prenatal and preimplantation genetic testing may be considered.
Cardioacrofacial dysplasia 1: a case report and literature review.
Cardioacrofacial dysplasia 1 [CAFD1; Online Mendelian Inheritance in Man (OMIM): #619142] is a rare skeletal ciliopathy caused by pathogenic variants in the PRKACA gene, exhibiting phenotypic overlap with conditions such as Ellis-van Creveld (EvC) syndrome. To date, only five cases have been reported worldwide, all carrying the identical p. Gly137Arg mutation. A 10-year-old male patient presented with short stature, progressive bilateral knee deformities, post-axial posterior polydactyly, and hypoplasia of teeth and nails since infancy. He had a history of partial atrial septal defect, functional single atrium, and pulmonary valve stenosis, undergoing cardiac repair at age 5 and bilateral polydactyly resection at age 7. Whole-exome sequencing (WES) confirmed a de novo heterozygous mutation in the PRKACA gene: c.409G>A (p.Gly137Arg). At age 10, the patient underwent robot-assisted bilateral proximal tibial epiphyseal fixation. One-month postoperative follow-up demonstrated significant improvement in gait and mobility. To our knowledge, this expands the known geographic distribution with PRKACA c.409G>A (p.Gly137Arg). The finding adds to prior reports that repeatedly implicate this variant; broader ascertainment is needed to establish whether it represents a true hotspot. In patients with an EvC-like phenotype who test negative for EVC/EVC2, screening of PRKACA can be considered. Prior work suggests that increased protein kinase A (PKA) catalytic activity may dampen Hedgehog (Hh) signaling, providing a plausible mechanism for the skeletal and cardiac findings. Early molecular diagnosis facilitates multidisciplinary management and genetic counseling.
Expanding the Coffin-Siris syndrome spectrum: genetic, dysmorphic, and endocrine findings in eight cases.
This study aims to expand the spectrum of Coffin-Siris syndrome (CSS), a rare and heterogeneous disorder, by thoroughly discussing its genetic, dysmorphic, and endocrine features through new cases and contributing to the literature. Eight patients who were referred to the genetics clinic with various complaints and subsequently diagnosed with CSS through microarray or clinical exome sequencing analyses were included in the study. The dysmorphic, genetic, and endocrine characteristics of eight genetically confirmed patients were evaluated. The patients, aged between 5 months and 6 years at the time of referral, comprised four females and four males. The most common reasons for referral were developmental delay and dysmorphic features. All patients exhibited varying degrees of dysmorphic facial features. Hypertrichosis, a typical feature of the syndrome, was present in five patients. Another characteristic finding was mild hypoplasia of the terminal fifth phalanges, observed in patients 1, 2, and 6. Consistent with this, mild/subtle hypoplasia and/or slight positional changes of the fifth fingernails were noted in these patients, rather than overt nail anomalies. In our study, eight variants were identified, two of which were novel. In our cohort, pathological short stature was observed in three patients, while hypothyroidism, transient hypercalcemia, cryptorchidism, and recurrent fractures were each identified in one patient. All three patients with short stature had delayed bone age with head circumference and BMI < - 2 SDS. Seven patients were diagnosed with ARID1B-related CSS type 1, while one patient was diagnosed with SMARCA4-related CSS type 4. Among the eight findings across patients, two were deletion-type copy-number variations (CNVs) identified by microarray analysis, and six were sequence variants: two frameshift, two splice-site, one nonsense, and one synonymous. Seven variants were classified as pathogenic and one as likely pathogenic. Family studies confirmed that the variants were de novo and validated their clinical relevance. CSS is a clinically and genetically heterogeneous syndrome. Patients may present with highly variable features, and typical signs of the syndrome may not be observed in all cases. This study expands the clinical spectrum of this rare syndrome and contributes to its genetic spectrum with the identification of new variants. • Coffin-Siris syndrome (CSS) is a clinically and genetically heterogeneous neurodevelopmental disorder most commonly caused by variants in SWI/SNF (BAF) complex genes (e.g., ARID1B, SMARCA4) and characterized by dysmorphic features, developmental delay, hypertrichosis, and fifth-digit/nail anomalies. • Endocrine and growth-related manifestations can occur in CSS, but their frequency and phenotypic range vary across cohorts and require individualized clinical follow-up. • This case series of eight genetically confirmed CSS patients (7 ARID1B, 1 SMARCA4) expands the phenotypic spectrum by detailing dysmorphic findings together with endocrine features including pathological short stature with delayed bone age, hypothyroidism, transient hypercalcemia, cryptorchidism, and recurrent fractures. • We identified eight pathogenic/likely pathogenic variants, including two novel variants, and highlight that fifth digit/nail involvement may be subtle (mild terminal fifth phalanx hypoplasia and minor fifth nail changes) rather than overt.
Publicações recentes
Retrospective longitudinal study on the long-term impact of COVID-19 infection on polysomnographic evaluation in patients with Prader-Willi syndrome.
French recommendations for the management of Behçet's disease.
📖 RevisãoDIGNiFI: Discovering causative genes for orphan diseases using protein-protein interaction networks.
Neurological outcome of patients with cryopyrin-associated periodic syndrome (CAPS).
📚 EuropePMCmostrando 200
Diets-Jongmans Syndrome due to a Novel KDM3B Variant: The First Molecularly Confirmed Case from Turkey.
Molecular syndromologyDiagnoses Live in Relationships: Bedside Sense in the Age of Precision Medicine.
Annals of family medicinePredictive Factors of Low HDL-C in Patients with Schizophrenia.
Neuropsychiatric disease and treatmentComplex Genetic Architecture in RASopathies: Constitutional PTPN11 and Mosaic RIT1 Pathogenic Variants Underlying Severe Noonan Syndrome With Adult-Onset Acute Myeloid Leukemia.
American journal of medical genetics. Part AHypertrophic Cardiomyopathy as a Key Feature of MRAS-Related Noonan Syndrome: New Case and Comprehensive Literature Review.
Prenatal diagnosisThe pathogenic ADAMTSL2 D167N variant causes geleophysic dysplasia-like connective tissue changes in mice.
The American journal of pathologyGender differences in the association between TyG-related index, metabolic score for insulin resistance, and overactive bladder: A cross-sectional study.
MedicineAnthropometric Measurements to Predict Metabolic Syndrome in Thai Women With Polycystic Ovary Syndrome: A Retrospective Study.
The journal of obstetrics and gynaecology researchAdipose tissue distribution and metabolic profile of young individuals with turner syndrome.
Journal of the Endocrine SocietyShort Stature and Growth Hormone Deficiency in POMC Deficiency: An Unexpected Clinical Association.
Journal of clinical research in pediatric endocrinologyNew insights into Oliver-McFarlane syndrome: adrenocortical hypofunction and variable expressivity in a Chinese sibling pair.
Journal of pediatric endocrinology & metabolism : JPEMPhosphoproteomics elucidates the functional impact of the PTPN11 p.Asn308Ser variant in a Noonan syndrome pedigree.
Journal of chromatography. B, Analytical technologies in the biomedical and life sciencesA rare case of severe short stature diagnosed after late-onset hypocalcemia: Kenny-Caffey syndrome type 2.
Journal of pediatric endocrinology & metabolism : JPEMCase Report: Compound heterozygous mutations in the IDUA gene causing mucopolysaccharidosis type I with uterine developmental abnormality.
Frontiers in pediatricsIron Deficiency Anemia Among Pediatric Celiac Disease Patients at the Armed Forces Hospital Southern Region: Prevalence, Predictors, and Outcomes.
CureusTherapy for Myhre Syndrome: Goals, Misconceptions, and Current Agents.
American journal of medical genetics. Part C, Seminars in medical geneticsPrecocious puberty: An overview of pathogenesis, clinical presentation, and management.
Best practice & research. Clinical obstetrics & gynaecologyA Diagnostic Dilemma: Concurrent Diagnosis of Cystic Fibrosis and Definitive Kabuki Syndrome Type 1.
International journal of molecular sciencesBody roundness index outperforms traditional obesity metrics in predicting cardiometabolic risk among children and adolescents: the EMSNGS study.
Frontiers in nutritionClinical significance of low-frequency mosaic thresholds (45,X/46,XX/47,XXX) in adulthood: A case report.
Taiwanese journal of obstetrics & gynecology[Genetic analysis of a Chinese pedigree affected with Isolated growth hormone deficiency due to variant of CHRHR gene].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsCardioacrofacial dysplasia 1: a case report and literature review.
Translational pediatricsThe epidemiology of cubital tunnel syndrome: a UK Biobank case-control study.
The Journal of hand surgery, European volumeObesity indices as predictors of metabolic syndrome: 1-year findings from a Peruvian cohort of private educational institution workers.
Therapeutic advances in endocrinology and metabolismNovel RAD50 variants lead to Nijmegen Breakage Syndrome-like disorder and unplanned recombinant human growth hormone treatment response.
Frontiers in endocrinologyExpanding the Coffin-Siris syndrome spectrum: genetic, dysmorphic, and endocrine findings in eight cases.
European journal of pediatricsLung-protective mechanical ventilation is not sex neutral.
British journal of anaesthesiaLamb-Shaffer syndrome in a Chinese adolescent: A case report.
MedicineThe Associations of Anthropometric Indices With Stages and Mortality in Cardiovascular-Kidney-Metabolic Syndrome: Insights From NHANES.
Reviews in cardiovascular medicineExploring the clinical and genetic spectrum of Steel syndrome: two case reports and review of the literature.
Frontiers in medicineGrip and pinch strengths and its association with cardiometabolic risk in children and adolescents aged 6 to 17 years.
Frontiers in nutritionA New Mathematical Model to Index Body Weight in Healthy Chinese Han Adults.
MedCommSPIN4-related X-linked overgrowth in a family.
European journal of medical geneticsA systems perspective on rare diseases: integrating human phenotype ontology with the Anukta framework of Ayurveda.
Journal of Ayurveda and integrative medicineIntegrating machine learning for advanced analysis of bioelectrical impedance parameters in children with nephrotic syndrome.
Frontiers in pediatricsAutism Spectrum Disorder in a Child with Floating-Harbor Syndrome: A Case Report.
Noro psikiyatri arsiviOnce-weekly somapacitan in children with Noonan syndrome: randomized controlled phase 3 trial.
European journal of endocrinologyMyhre Syndrome Presenting With Congenital Proximal Radioulnar Synostosis: A Case Report.
CureusA novel variant in ARID2 causes Coffin-Siris syndrome 6 with liver cirrhosis.
GeneDNMT3A R882C variant in a patient with a presumed pineal gland tumor, highlighting potential tumor susceptibility in Tatton-Brown-Rahman syndrome.
Cancer geneticsLong-Term Outcomes of Successful Treatment with Ruxolitinib in a Pediatric Patient with Autoimmune Lymphoproliferative Syndrome and a Signal Transducer and Activator of Transcription 3 Gain-of-Function Mutation.
Klinische PadiatrieAssociation between Body Roundness Index and infertility risk in Han Chinese women: a retrospective observational study.
Frontiers in medicineThe Cut-Off for Triglyceride-Glucose-Body Mass Index (TyG-BMI) and Triglyceride-Glucose-Waist Circumference Index (TyG-WC) Discriminating the Insulin Resistance Based on the SHBG Level and HOMA-IR Value in Caucasian Women with Polycystic Ovary Syndrome.
Medicina (Kaunas, Lithuania)Effect of Oral Glucose Administration on Ghrelin Levels in Normal-Height Prepubertal Children Born Small for Gestational Age (SGA).
International journal of molecular sciencesPhenotypic Variability Associated with Jagunal Homolog 1 (JAGN1) Deficiency Caused by the c.63G>T Variant.
International journal of molecular sciencesXp22.33 Duplication Encompassing PAR1 in a Male with Syndromic Neurodevelopmental Disorder and Tall Stature.
GenesEmerging role of KDM5C in X-linked intellectual disability based on human genetic data and zebrafish models.
Frontiers in molecular neuroscienceRefractory Rickets: Evaluation and Management.
Indian journal of pediatricsFamilial pneumothorax in twins with Tatton-Brown-Rahman DNMT3A overgrowth syndrome.
European journal of human genetics : EJHGA Novel Association Between Mandibulofacial Dysostosis with Microcephaly and Congenital Diaphragmatic Hernia.
The Cleft palate-craniofacial journal : official publication of the American Cleft Palate-Craniofacial AssociationNutrient and endocrine factors affecting impaired growth in pediatric mitochondrial diseases.
Endocrinology, diabetes & metabolism case reports[Risk factor analysis of excessive resection of facet joint after large-channel spinal endoscopic depression for degenerative lumbar diseases].
Zhongguo xiu fu chong jian wai ke za zhi = Zhongguo xiufu chongjian waike zazhi = Chinese journal of reparative and reconstructive surgeryAssociation between body roundness index and reproductive outcomes in patients with polycystic ovary syndrome: a secondary analysis based on PCOSAct.
Frontiers in nutritionFamilial cartilage-hair hypoplasia: prenatal ultrasound features and clinical outcomes in three siblings with identical RMRP variants.
BMJ case reportsMetabolic Risk Assessment Using the Triglyceride-Glucose Index in Patients with Adrenal Insufficiency on Glucocorticoid Therapy.
Metabolic syndrome and related disordersAssessing the diagnostic ability of adiposity measures to identify cardiometabolic risk factors and the metabolic syndrome in South African corporate employees.
Cardiovascular journal of AfricaNeuronal Heterotopy in a Patient with Wiedemann-Steiner Syndrome Caused by a Truncating KMT2A Variant: Clinical and Genetic Correlations.
Reports (MDPI)Childhood Obesity: A Multisystem Challenge Linking Hypertension, NAFLD, and Sleep Apnea.
Medical sciences (Basel, Switzerland)A radiological case series of three siblings with osteogenesis imperfecta and shared paternal inheritance.
Radiology case reportsEffectiveness of body roundness index, relative fat mass, and body adiposity index in predicting body adiposity, insulin resistance, and metabolic syndrome in women.
Nutrition (Burbank, Los Angeles County, Calif.)Exome findings in children with short stature evaluated by growth hormone stimulation testing.
European journal of endocrinologyClinical features and management of 16q24.3 microdeletion KBG syndrome: literature review.
Frontiers in pediatricsNovel ocular feature in oculoskeletodental syndrome: high axial myopia and megalocornea in a child with a homozygous PIK3C2A variant.
Ophthalmic geneticsA novel CEP57 gene mutation in mosaic variegated aneuploidy syndrome 2: case report.
Journal of pediatric endocrinology & metabolism : JPEMAssessment of combined growth hormone and gonadotropin-releasing hormone analogue treatment in children with Silver-Russell syndrome.
Hormone research in paediatricsEstimation of Transpulmonary Driving Pressure, Strain, and Lung-Specific Elastance Using Volumetric Methods in Invasively Ventilated Children-A Feasibility Study.
Pediatric critical care medicine : a journal of the Society of Critical Care Medicine and the World Federation of Pediatric Intensive and Critical Care SocietiesDiagnostic odyssey of a male with 45,X/46,XY mosaicism: case report and review of the literature.
F&S reportsLong-term Oral Management for 2q37 Deletion Syndrome Patient.
The Bulletin of Tokyo Dental CollegeAssociation of Cardiometabolic Index with Mortality Risk in Early-Stage Cardiovascular-Kidney-Metabolic Syndrome and Its Potential Role in Metabolically Unhealthy Phenotypes: Evidence from NHANES 2007-2018.
Metabolic syndrome and related disordersPhenotype Variations in a Family with Various Rearrangements in the Locus of the SHOX Gene.
International journal of molecular sciencesDeterminants of Health-Related Quality of Life in Women with Turner Syndrome: The Role of Comorbidities, Hormonal Therapy and Depressive Symptoms.
Journal of clinical medicineExpanding the Evaluation of Skeletal Anomalies in Patients With KBG Syndrome: Recommendations for Clinical Practice.
American journal of medical genetics. Part AIntegrative metabolomics and proteomics reveal early cardiovascular risk signatures in PCOS female offspring.
Journal of ovarian researchHigher skin carotenoid levels are associated with lower risks of metabolic syndrome: a cross-sectional study in Vietnamese participants.
Frontiers in nutritionMETS-VF outperforms traditional adiposity indices in predicting overactive bladder risk: a NHANES-based cross-sectional study.
Translational andrology and urologyUnraveling the Mechanistic Spectrum of Myhre Syndrome: SMAD4 Signaling Disruption, Skeletal Phenotypes, and Translational Innovation.
American journal of medical genetics. Part C, Seminars in medical geneticsGrowth patterns: Pathology vs. Normal variation.
Growth hormone & IGF research : official journal of the Growth Hormone Research Society and the International IGF Research SocietyClinical Presentation and Management of Swyer Syndrome: A Case Report.
CureusPrevalence of sarcopenia in patients with chronic intestinal failure due to short bowel syndrome.
Intestinal Failure (New York, N.Y.)Cerebral Calcification and Treatment-Resistant Seizures: A Rare Syndromic Presentation of Pseudohypoparathyroidism-A Case Report.
AACE endocrinology and diabetesA novel homozygous ADAMTS10 frameshift variant in Weill-Marchesani syndrome in a Chinese family.
BMC medical genomicsAnalysis of metabolic status and risk factors of small for gestational age children with catch-up growth in East China.
BMC pediatricsValidity and reliability of a scale of activities of daily living at home in children, youth and adults with Down syndrome in Chile.
Journal of health, population, and nutritionLower Genetically Predicted Circulating Insulin-like Growth Factor-1 is Associated with a Higher Risk of Adolescent Idiopathic Scoliosis: A Mendelian Randomization Study.
SpinePPP1R12A Mutation Presenting With Congenital Jejunal Atresia and Short Stature: A Pediatric Endocrinology Case Report.
Case reports in pediatricsThe Metacarpophalangeal Pattern Profile: An Old Method With New Insights Into the Evaluation of Short Stature.
American journal of human biology : the official journal of the Human Biology CouncilA novel de Novo KCNC1 mutation (c.1147 C > T) presenting with epilepsy and ADHD: a case report and literature review.
BMC neurologyAn examination of child outcomes and predictors of severity when prenatal alcohol exposure is frequent and heavy.
Alcohol, clinical & experimental researchClinical characterization and analysis of the POLE gene in three Chinese patients with IMAGEI syndrome.
BMC pediatricsLongitudinal functional lung imaging in children with Post-COVID-19 syndrome.
Molecular and cellular pediatricsTriglyceride-glucose index and triglyceride-glucose index-related markers for identifying metabolic syndrome in Saudi Arabia.
Journal of clinical lipidologyAllogeneic Hematopoietic Cell Transplantation for Morquio A Syndrome: An International Retrospective Study.
Transplantation and cellular therapyNeonatal erythroderma and immunodysplasia: Overlap of cartilage-hair hypoplasia and Omenn syndrome.
European journal of medical geneticsMeier-Gorlin syndrome due to a recurrent DONSON variant in a Turkish family: first report of thumb aplasia and long-term growth data.
Journal of pediatric endocrinology & metabolism : JPEMA Case of FAM111A-Associated Kenny-Caffey Syndrome Type 2 with New Clinical Features: Microtia, Lacunar Skull Appearance, and Arnold-Chiari Malformation.
Molecular syndromologyAssociations Between Serum 25-Hydroxyvitamin D Levels and Metabolic Syndrome Among Korean Adolescents: Based on the Korea National Health and Nutrition Examination Survey in 2022-2023.
NutrientsDirections and Perspectives for Preventive Activities in Primary Care-Patients' Health-Promoting and Health-Risk Behaviours.
NutrientsSkin Carotenoid Score as a Potential Early Biomarker of Metabolic Syndrome Risk in Adolescents.
NutrientsReal-World Evidence of Growth Improvement in Children 1 to 5 Years of Age Receiving Enteral Formula Administered Through an Immobilized Lipase Cartridge.
NutrientsBeyond Neurodevelopmental Delay: BICRA-Related Coffin-Siris Syndrome 12 with Severe Intestinal Dysmotility and Recurrent Pneumothorax.
GenesPrenatal Diagnosis of a Feingold Syndrome Pregnancy Complicated with Severe Preeclampsia: A Report of a Challenging Case.
GenesArtificial Neural Network as a Tool to Predict Severe Toxicity of Anticancer Drug Therapy in Patients with Gastric Cancer: A Retrospective Study.
Diagnostics (Basel, Switzerland)Some Biomechanical and Anthropmetric Differences Between Elite Swimmers with Down Syndrome and Intellectual Disabilities.
Sports (Basel, Switzerland)A Rare Co-occurrence of Duchenne Muscular Dystrophy and Glycerol Kinase Deficiency Associated With Xp21 Contiguous Gene Deletion Syndrome: A Case Report.
CureusEffectiveness of a nursing model based on traditional Chinese medicine syndrome differentiation in patients with type 2 diabetes: A retrospective cohort study.
MedicineNoonan syndrome spectrum disorders in real life: patient characteristics and response to growth hormone therapy in a genetically defined single-country multicenter cohort.
European journal of pediatricsEfficacy of Recombinant Human Growth Hormone on Glucocorticoid-Induced Short Stature in Children: A Retrospective Controlled Study.
Hormone research in paediatricsA novel compound heterozygous mutation in ADAMTS17 identified in a Chinese family with Weill-Marchesani syndrome.
International journal of ophthalmologyMapping the Prenatal Growth of the Mandible.
The Journal of craniofacial surgeryImpact of using waist-to-height ratio for diagnosing obesity in general digestive outpatient consultations.
Revista espanola de enfermedades digestivasProspective associations of triglyceride-glucose related indices with cardiovascular disease and mortality in individuals with metabolic syndrome: evidence from the UK biobank.
Cardiovascular diabetologySevere Short Stature, Pathological Fracture, and Avascular Necrosis Due to Prolonged Over-the-Counter Steroid Use in an Adolescent Patient.
CureusNovel variants in STAG2 and PKD1 associate with multiple congenital malformations and autosomal dominant polycystic kidney disease in a Chinese family: A case report and literature review.
Experimental and therapeutic medicineMauriac Syndrome: Growth and Clinical Outcomes After 2.5 Years of Automated Insulin Delivery Treatment.
JCEM case reportsAdvanced-Stage Gonadal Dysgerminoma in a Patient With a Previous Diagnosis of Familial Swyer Syndrome: A Very Rare Genetic Entity.
Case reports in medicineA Novel Evaluation of 24-hour Energy Metabolism in Cushing's Syndrome: The Metabolic Cost of Hypercortisolism.
Journal of the Endocrine SocietyDyggve-Melchior-Clausen syndrome in three siblings: a unique case series with dual diagnosis of Down syndrome and Hirschsprung disease.
Journal of pediatric endocrinology & metabolism : JPEMAssessment of Nutrition Quality in People With Prader-Willi Syndrome in Australia.
Journal of human nutrition and dietetics : the official journal of the British Dietetic AssociationPrognostic effect of triglyceride glucose-related parameters on all-cause and cardiovascular mortality in individuals with cardiovascular-kidney-metabolic syndrome: evidence from international multi-cohort studies.
Cardiovascular diabetologyAnalysis of dry eye syndrome incidence and prognostic factors in 778 patients following refractive corneal surgery.
Photodiagnosis and photodynamic therapyPrenatal Shwachman-Diamond Syndrome: Diagnostic Challenges in Two Unrelated Cases With a Rare Clinical Presentation and Pseudogene Interference, and a Review of the Literature.
Prenatal diagnosisInternational guideline on genetic testing of children with short stature.
European journal of endocrinologyDiagnostic yield of genetic testing in children with short stature: a systematic review.
European journal of endocrinologyStructural instability impairs function of the UDP-xylose synthase 1 Ile181Asn variant associated with short-stature genetic syndrome in humans.
FEBS lettersA study on the nutritional status and body composition of children with Hutchinson-Gilford progeria syndrome.
Orphanet journal of rare diseasesClinical Anthropometry in Male Infertility: Differences Between Men With and Without Klinefelter Syndrome.
CureusNovel characterization of MRAS mutation-associated Noonan syndrome: Mild adult-onset hypertrophic cardiomyopathy combined with infective endocarditis: A case report.
MedicineNeuropeptides and the Autonomic Nervous System in Prader-Willi Syndrome.
International journal of molecular sciencesEvaluation of wrist radiographic indices in people with idiopathic carpal tunnel syndrome: a prospective cross-sectional study.
Annals of physical and rehabilitation medicineTriglyceride-glucose-related parameters cutoff points for predicting the prevalence of metabolic syndrome in elderly Caucasian patients with obesity.
Nutricion hospitalariaThe Association Between Height-Related Novel Anthropometric Indices and Cardiovascular Events: A Cross-Sectional Insight From the MASHAD Cohort Study.
Health science reportsImpact of growth hormone treatment on a 12-year-old female with newly diagnosed panhypopituitarism and distal arthrogryposis.
Endocrinology, diabetes & metabolism case reportsComparative predictive value of anthropometric indexes for hypertension: a 15-year prospective cohort study.
Internal and emergency medicineIdentification of an unusual variant of Fanconi-Bickel syndrome presenting as proximal tubulopathy and short stature.
BMJ case reportsLong-Term experience with growth hormone therapy in pediatric growth disorders: an analysis of the LG growth study data.
EndocrineCase Reports: Exploring the Varied Presentations and Clinical Features of Carney Complex, A Detailed Report on Three Distinct Cases.
Journal of clinical research in pediatric endocrinologyNovel variant in FGFR2 in a family with anterior segment anomalies.
Ophthalmic geneticsPediatric floating-harbor syndrome: clinical features and treatment outcomes in a cohort of Chinese children.
European journal of pediatricsDual-Genetic Etiology in an Atypical Dent Disease Phenotype Which Combines Features of Focal Segmental Glomerulosclerosis and Ellis-Van Creveld-Like Syndrome: A Case Report.
Case reports in nephrology and dialysisVentricular arrhythmia and Noonan syndrome with leucine zipperlike transcription regulator 1 mutations: expanding the phenotype with a case report and review of the literature.
Cardiology in the youngGrowth Hormone Therapy in 5q35 Duplication Syndrome: Evidence From a Long-Term Follow-Up.
Clinical endocrinologyDXA Derived Low Bone Mass in a Cohort of Prepubertal Eastern Indian Girls With Turner Syndrome Disappeared Following Adjustment for Short Stature.
Clinical endocrinologyDe novo variants in KDM2A cause a syndromic neurodevelopmental disorder.
American journal of human geneticsCardiovascular phenotypes of children and adolescents with Turner syndrome from a single-center cohort study.
Orphanet journal of rare diseasesGenetic Heterogeneity Underlying Familial Short Stature.
Diagnostics (Basel, Switzerland)Identification of a Novel Nonsense Mutation in the IGSF1 Gene Reveals Sex-Specific Phenotypic Variability Within a Single Family.
Children (Basel, Switzerland)Bone health and body composition in Indian girls with Turner syndrome: analysis based on therapeutic interventions.
EndocrineDiabetes mellitus in SHORT syndrome managed with multi-agent oral therapies: a case report and literature review.
Therapeutic advances in endocrinology and metabolismGenotype-Phenotype Correlations in Klinefelter and Turner Syndrome: A Decade of Sex Chromosome Aneuploidy Data From a Single Academic Medical Center.
Molecular genetics & genomic medicineTeduglutide in pediatric patients under 10 kg with short bowel syndrome on parenteral support: An open-label study.
Pediatrics international : official journal of the Japan Pediatric SocietyTRISOMY 21: development of syndrome-specific growth charts from a wide Italian cohort and validation of BMI as a predictor of increased risk of metabolic derangement.
Journal of endocrinological investigation[Clinical and genetic analysis of a Chinese pedigree affected with Vissers-Bodmer syndrome due to variant of CNOT1 gene and a literature review.].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsBathrocephaly and Serpentine Fibula as Underrated Features of Osteogenesis Imperfecta Type I: A Case Report.
Molecular syndromologyAssociation Between Novel Inflammatory Indices and Metabolic Syndrome in Children and Adolescents with Obesity.
Metabolic syndrome and related disordersDiagnostic Accuracy of the Triglyceride-Glucose Derived Indices in Detecting Metabolic Syndrome in Pediatric Patients.
Journal of obesity & metabolic syndromeGenetic activation of ERK2 recapitulates core neurodevelopmental features of Rasopathy syndromes in mice.
bioRxiv : the preprint server for biologyA pediatric case of diphthamide biosynthesis 1 gene defect presenting with developmental delay, short stature, dysmorphic features, and sparse hair (Loucks-Innes syndrome): a case report.
Journal of medical case reportsUltrasound Assessment of the Tibial Nerve at the Retromalleolar Level: Influence of Anthropometric Characteristics and Clinical Implications.
Clinics and practiceCase Report of a Novel EVC Gene Mutation in Ellis-van Creveld Syndrome: Implications for Pediatric Dental Management.
Case reports in dentistry3-M syndrome: evolution of the phenotype over time.
Italian journal of pediatricsComparison of novel and traditional anthropometric indices: which is the best indicator of frailty in older adults?
Aging clinical and experimental researchPrevalence and correlates of overweight and obesity in patients with major depression accompanied by abnormal TSH levels.
BMC psychiatryHeterozygous loss of OSR2 can cause radioulnar synostosis with ancillary skeletal manifestations.
Genetics in medicine : official journal of the American College of Medical GeneticsReal-World Evidence of Treatment Patterns and Costs of Turner Syndrome and Noonan Syndrome in the USA.
Hormone research in paediatricsResting Energy Expenditure in Adults With Williams Syndrome: Comparative Accuracy of Predictive Equations.
Journal of intellectual disability research : JIDRNovel KDM3B Variants in Two Chinese Patients With Global Developmental Delay and Autism.
International journal of developmental neuroscience : the official journal of the International Society for Developmental NeuroscienceC-type natriuretic peptide as mediator of growth in the absence of growth hormone: Unraveling the mystery of the growth without GH syndrome.
Hormones (Athens, Greece)A Case of CSNK2A1 Gene Variant Causing Okur-Chung Syndrome and Analysis of the Clinical Phenotypic Spectrum.
Molecular genetics & genomic medicinePhenotypic Analysis of Embryos in a Noonan Syndrome Model Mouse With the Rit1 A57G Mutation.
Molecular genetics & genomic medicineTatton-Brown-Rahman Syndrome Due to a Novel DNMT3A Variant Presenting With Autism, Attention-Deficit/Hyperactivity Disorder (ADHD), and Regression: A Saudi Case Report.
CureusAnalysing concordance between MUAC, MUACZ, and WHZ in diagnosing acute malnutrition among children under five in Somalia.
Journal of global health[Selection of compression-distraction device type for mandibular hypoplasia based on mandible morphometric parameters].
StomatologiiaChildren and adolescents with DiGeorge syndrome are short in early infancy but develop excess weight in adolescence: a retrospective study.
Revista paulista de pediatria : orgao oficial da Sociedade de Pediatria de Sao PauloSuccessful kidney transplantation in a patient with late-onset type II Bartter syndrome: A rare case report.
Clinical nephrology. Case studiesHeterogeneity of Orodental Features in a Family with Noonan Syndrome.
International journal of molecular sciencesComparative analysis of the bone age of wrist bones in Chinese children with different causes of short stature and central precocious puberty.
Quantitative imaging in medicine and surgeryFurther evidence for a wide phenotypic and mutational spectrum of Cohen syndrome: case report and literature review.
Journal of applied geneticsAdolescent "Lean PCOS" Is Characterized by Higher Insulin Resistance and Adverse Adipokine Profile.
The Journal of clinical endocrinology and metabolismClinical and Genotypic Insights into Turner Syndrome: Emphasis on Cardiovascular Abnormalities.
Journal of the ASEAN Federation of Endocrine SocietiesEpidural Anesthesia in a Patient With Turner Syndrome: A Case Report.
CureusAssociation of adiposity indices with insulin resistance among women with polycystic ovary syndrome.
Clinical and experimental reproductive medicineAssociations between exposure to environmental pollutants, metabolic syndrome risk, and obesity-related anthropometric indices.
International journal of hygiene and environmental healthClinical outcomes of exclusive enzyme therapy (laronidase) in a cohort of patients with mucopolysaccharidosis type I.
Orphanet journal of rare diseasesWeight-adjusted waist index outperforms other obesity indices for cardiovascular disease prediction in cardiovascular-kidney-metabolic syndrome: insights from UK biobank.
BMC public healthGrowth and sleep outcomes after adenotonsillectomy in pediatric mild sleep-disordered breathing.
Scientific reportsBurden of anthropometric failures and concordance of mid-upper arm circumference with weight for length z score in identifying malnutrition among under 2-year-old children in Southern India.
Public health nutritionMucopolysaccharidosis or Skeletal Dysplasia?: Important Clinical and Radiologic Clues for Differential Diagnosis of Based on Difficult Cases.
Journal of clinical research in pediatric endocrinologyShort stature, optic atrophy, and Pelger-Huët anomaly (SOPH) syndrome: report of a case lacking neutrophil morphologic changes and review of literature.
Ophthalmic geneticsAssociations of 13C-Sucrose Breath Test Dynamics with Anthropometry and Demographics: A Comparison of Studies in the United Kingdom and Zambia.
Current developments in nutritionPathogenic variants in the cohesin loader subunit MAU2 lead to a new Cornelia de Lange Syndrome subtype.
medRxiv : the preprint server for health sciencesPrevalence of physically active females at risk for the female athlete triad in Spain.
Journal of the International Society of Sports NutritionBody Roundness Index Versus Body Mass Index: Differential Associations With Obstructive Sleep Apnea Syndrome and All-Cause Mortality in US Adults Aged 20 Years and Older.
Brain and behaviorManaging acute myeloid leukemia in the context of sickle cell anemia and suspected Fanconi anemia in Tanzania: a case report.
Journal of medical case reportsTemporal Bone CT Findings in Hajdu-Cheney Syndrome: Case Report with Review of the Literature.
AJNR. American journal of neuroradiologyEndoCompass Project: Research Roadmap for Growth Disorders.
Hormone research in paediatricsIncreased risk for severe peripheral arterial disease in Kabuki syndrome.
Journal of vascular surgery cases and innovative techniquesHyperglycemic Hyperosmolar State as the Initial Presentation of Wolfram Syndrome: A Common Complication Revealing a Rare Disease-A Case Report.
Clinical case reportsSuccessful Use of Haploidentical HSCT in a Child With Schimke Immuno-Osseous Dysplasia Who Developed PTLD After Kidney Transplantation.
Pediatric transplantationDelayed Diagnosis of 48XXYY Syndrome: A Case Report Highlighting the Role of G-Banding Cytogenetics.
Journal of UOEHAssociation of body roundness index with cardiovascular disease in early Cardiovascular-Kidney-Metabolic syndrome stage 0-3: mediation by the triglyceride-glucose index in a national cohort study.
Diabetology & metabolic syndromeAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de ataxia-fotossensibilidade-baixa estatura.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de ataxia-fotossensibilidade-baixa estatura
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Phosphoproteomics elucidates the functional impact of the PTPN11 p.Asn308Ser variant in a Noonan syndrome pedigree.Journal of chromatography. B, Analytical technologies in the biomedical and life sciences· 2026· PMID 41843963mais citado
- Case Report: Compound heterozygous mutations in the IDUA gene causing mucopolysaccharidosis type I with uterine developmental abnormality.
- A Diagnostic Dilemma: Concurrent Diagnosis of Cystic Fibrosis and Definitive Kabuki Syndrome Type 1.
- Cardioacrofacial dysplasia 1: a case report and literature review.
- Expanding the Coffin-Siris syndrome spectrum: genetic, dysmorphic, and endocrine findings in eight cases.
- Retrospective longitudinal study on the long-term impact of COVID-19 infection on polysomnographic evaluation in patients with Prader-Willi syndrome.
- French recommendations for the management of Behçet's disease.
- DIGNiFI: Discovering causative genes for orphan diseases using protein-protein interaction networks.
- Neurological outcome of patients with cryopyrin-associated periodic syndrome (CAPS).
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1184(Orphanet)
- MONDO:0015248(MONDO)
- GARD:2287(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Q55785354(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome de ataxia-fotossensibilidade-baixa estatura
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata