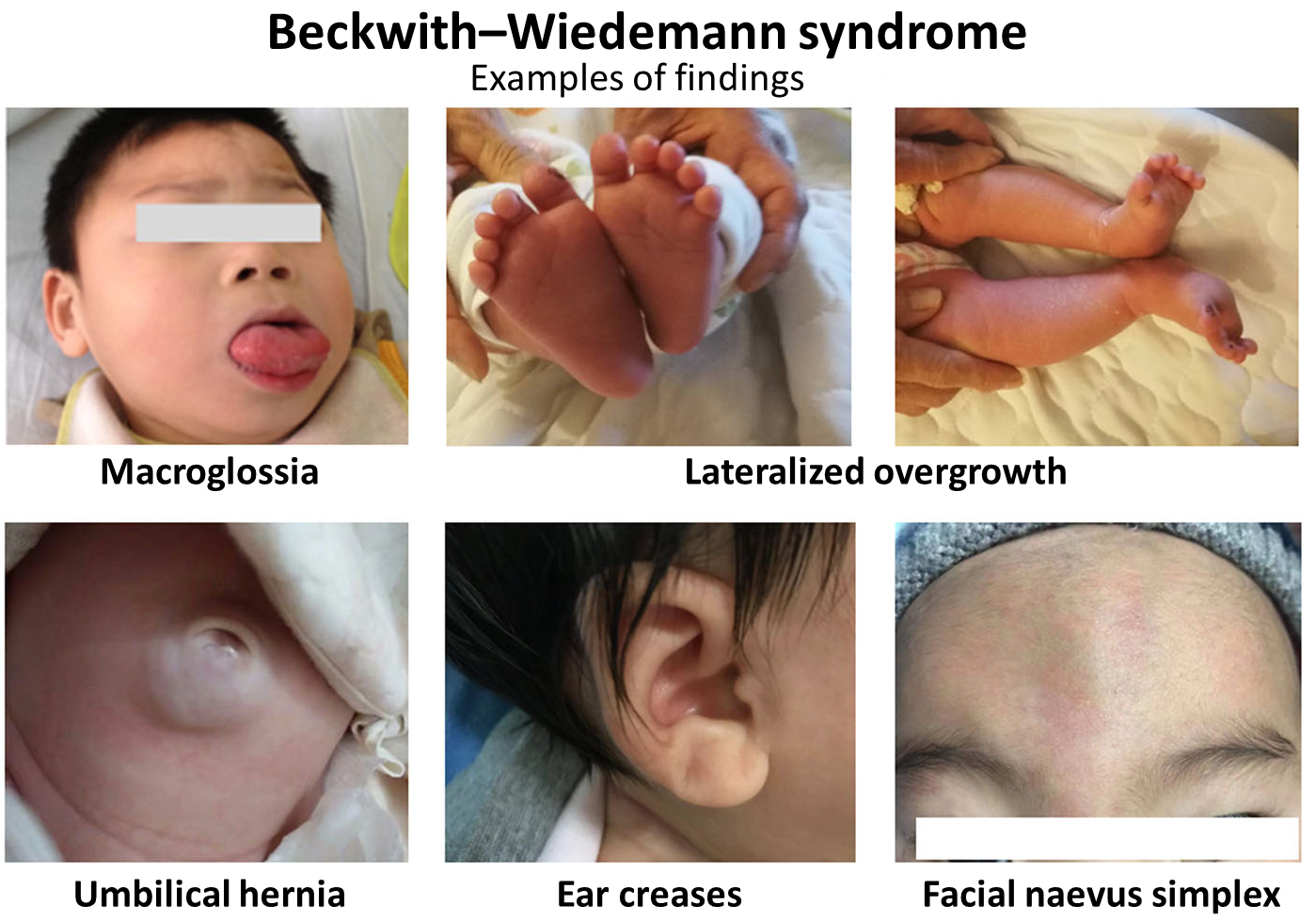
A síndrome de Simpson-Golabi-Behmel (SGBS) tipo 2 é uma forma da SGBS extremamente rara, grave e que leva à morte nos primeiros anos de vida. É uma síndrome que causa crescimento exagerado e várias anomalias, caracterizada por: inchaço generalizado no feto (hidropsia fetal), tamanho da cabeça maior que o normal (macrocefalia), características faciais incomuns como olhos muito separados (hipertelorismo), orelhas baixas e viradas para trás, nariz curto e largo com narinas viradas para cima, a área entre o nariz e o lábio superior mais marcada (filtro labial proeminente), boca grande com lábio superior fino, céu da boca em arco alto e fenda (fenda palatina); além de pescoço curto, pele solta/excessiva, defeitos nos ossos (afetando braços e pernas), unhas pequenas e pouco desenvolvidas, alterações nos sistemas digestório e geniturinário, baixo tônus muscular (hipotonia) e comprometimento neurológico. Deficiência intelectual grave, obesidade e infecções (como pneumonia e sepse, uma infecção generalizada) também foram relatadas.
Introdução
O que você precisa saber de cara
A síndrome de Simpson-Golabi-Behmel (SGBS) tipo 2 é uma forma da SGBS extremamente rara, grave e que leva à morte nos primeiros anos de vida. É uma síndrome que causa crescimento exagerado e várias anomalias, caracterizada por: inchaço generalizado no feto (hidropsia fetal), tamanho da cabeça maior que o normal (macrocefalia), características faciais incomuns como olhos muito separados (hipertelorismo), orelhas baixas e viradas para trás, nariz curto e largo com narinas viradas para cima, a área entre o nariz e o lábio superior mais marcada (filtro labial proeminente), boca grande com lábio superior fino, céu da boca em arco alto e fenda (fenda palatina); além de pescoço curto, pele solta/excessiva, defeitos nos ossos (afetando braços e pernas), unhas pequenas e pouco desenvolvidas, alterações nos sistemas digestório e geniturinário, baixo tônus muscular (hipotonia) e comprometimento neurológico. Deficiência intelectual grave, obesidade e infecções (como pneumonia e sepse, uma infecção generalizada) também foram relatadas.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 3 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 19 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição.
Component of the centrioles controlling mother and daughter centrioles length. Recruits to the centriole IFT88 and centriole distal appendage-specific proteins including CEP164 (By similarity). Involved in the biogenesis of the cilium, a centriole-associated function. The cilium is a cell surface projection found in many vertebrate cells required to transduce signals important for development and tissue homeostasis (PubMed:33934390). Plays an important role in development by regulating Wnt signa
Cytoplasm, cytoskeleton, microtubule organizing center, centrosome, centrioleCytoplasm, cytoskeleton, cilium basal bodyNucleusCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centriolar satellite
Orofaciodigital syndrome 1
A form of orofaciodigital syndrome, a group of heterogeneous disorders characterized by abnormalities in the oral cavity, face, and digits and associated phenotypic abnormalities that lead to the delineation of various subtypes. OFD1 is X-linked dominant syndrome, lethal in males. Craniofacial findings consist of facial asymmetry, hypertelorism, median cleft, or pseudocleft of the upper lip, hypoplasia of the alae nasi, oral clefts and abnormal frenulea, tongue anomalies (clefting, cysts, hamartoma), and anomalous dentition involving missing or extra teeth. Asymmetric brachydactyly and/or syndactyly of the fingers and toes occur frequently. Approximately 50% of OFD1 females have some degree of intellectual disability. Some patients have structural central nervous system anomalies such as agenesis of the corpus callosum, cerebellar agenesis, or a Dandy-Walker malformation. Patients with OFD1 can develop fibrocystic disease of the liver and pancreas, in addition to polycystic kidneys.
Variantes genéticas (ClinVar)
445 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 145 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
8 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de Simpson-Golabi-Behmel tipo 2
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Expanding the Genotypic and Phenotypic Spectrum of OFD1-Related Conditions: Three More Cases.
Pathogenic variants in the OFD1 gene are linked to a spectrum of syndromes that exhibit partial clinical overlap. Hemizygous loss-of-function variants are considered lethal in males, while heterozygous loss-of-function variants generally result in oro-facial-digital syndrome type 1. A reported phenotype, Simpson-Golabi-Behmel syndrome type 2, was published once but remains controversial, with many specialists questioning its validity and arguing about its continued listing in the OMIM database. To investigate the genetic and phenotypic characteristics of the patients, we performed clinical exome sequencing, family-based genetic analysis, X-inactivation studies, electron microscopy, and detailed clinical assessments. Three patients from unrelated families carrying loss-of-function variants in the OFD1 gene were identified, emphasizing the diverse phenotypic spectrum of OFD1-associated disorders. The first patient, a female with a heterozygous frameshift variant p.(Gln398LeufsTer2), was diagnosed with oro-facial-digital syndrome type 1. The second patient, a male with a heterozygous nonsense variant p.(Gln892Ter), presented with features resembling Simpson-Golabi-Behmel syndrome type 2, as previously reported under this diagnosis. The third patient, a male with another heterozygous nonsense variant p.(Glu879Ter), exhibited isolated primary ciliary dyskinesia without any syndromic features. This study contributes to the growing body of evidence on the expanding phenotypic spectrum of OFD1-associated disorders. It underscores the need for further investigation into the molecular mechanisms underlying the diverse presentations and the necessity of re-evaluating diagnostic classifications for conditions such as SGBS2 in the context of variants in the OFD1 gene.
OFD Type I syndrome: lessons learned from a rare ciliopathy.
The OFD1 gene was initially identified as the gene responsible for the X-linked dominant male lethal OFD type I syndrome, a developmental disorder ascribed to cilia disfunction. The transcript has been subsequently associated to four different X-linked recessive conditions, namely Joubert syndrome, retinitis pigmentosa, primary ciliary dyskinesia and Simpson-Golabi-Behmel type 2 syndrome. The centrosomal/basal body OFD1 protein has indeed been shown to be required for primary cilia formation and left-right asymmetry. The protein is also involved in other tasks, e.g. regulation of cellular protein content, constrain of the centriolar length, chromatin remodeling at DNA double strand breaks, control of protein quality balance and cell cycle progression, which might be mediated by non-ciliary activities. OFD1 represents a paradigmatic model of a protein that performs its diverse actions according to the cell needs and depending on the subcellular localization, the cell type/tissue and other possible factors still to be determined. An increased number of multitask protein, such as OFD1, may represent a partial explanation to human complexity, as compared with less complex organisms with an equal or slightly lower number of proteins.
The expanding phenotype of OFD1-related disorders: Hemizygous loss-of-function variants in three patients with primary ciliary dyskinesia.
OFD1 has long been recognized as the gene implicated in the classic dysmorphology syndrome, oral-facial-digital syndrome type I (OFDSI). Over time, pathogenic variants in OFD1 were found to be associated with X-linked intellectual disability, Joubert syndrome type 10 (JBTS10), Simpson-Golabi-Behmel syndrome type 2 (SGBS2), and retinitis pigmentosa. Recently, OFD1 pathogenic variants have been implicated in primary ciliary dyskinesia (PCD), a disorder of the motile cilia with a phenotype that includes recurrent oto-sino-pulmonary infections, situs abnormalities, and decreased fertility. We describe three male patients with PCD who were found to have hemizygous pathogenic variants in OFD1, further supporting that PCD is part of a clinical spectrum of OFD1-related disorders. In addition, we provide a review of the available clinical literature describing patients with OFD1 variants and highlight the phenotypic variability of OFD1-related disease. Some individuals with hemizygous OFD1 variants have PCD, either apparently isolated or in combination with other features of OFD1-related disorders. As clinicians consider the presence or absence of conditions allelic at OFD1, PCD should be considered part of the spectrum of OFD1-related disorders. Understanding the OFD1-related disease spectrum may allow for more focused genetic testing and more timely management of treatable sequelae.
Truncating mutations in exons 20 and 21 of OFD1 can cause primary ciliary dyskinesia without associated syndromic symptoms.
Primary ciliary dyskinesia (PCD) is a motile ciliopathy, whose symptoms include airway infections, male infertility and situs inversus. Apart from the typical forms of PCD, rare syndromic PCD forms exist. Mutations of the X-linked OFD1 gene cause several syndromic ciliopathies, including oral-facial-digital syndrome type 1, Joubert syndrome type 10 (JBTS10), and Simpson-Golabi-Behmel syndrome type 2, the latter causing the X-linked syndromic form of PCD. Neurological and skeletal symptoms are characteristic for these syndromes, with their severity depending on the location of the mutation within the gene. To elucidate the role of motile cilia defects in the respiratory phenotype of PCD patients with C-terminal OFD1 mutations. Whole-exome sequencing in a group of 120 Polish PCD patients, mutation screening of the OFD1 coding sequence, analysis of motile cilia, and magnetic resonance brain imaging. Four novel hemizygous OFD1 mutations, in exons 20 and 21, were found in men with a typical PCD presentation but without severe neurological, skeletal or renal symptoms characteristic for other OFD1-related syndromes. Magnetic resonance brain imaging in two patients did not show a molar tooth sign typical for JBTS10. Cilia in the respiratory epithelium were sparse, unusually long and displayed a defective motility pattern. Consistent with the literature, truncations of the C-terminal part of OFD1 (exons 16-22) almost invariably cause a respiratory phenotype (due to motile cilia defects) while their impact on the primary cilia function is limited. We suggest that exons 20-21 should be included in the panel for regular mutation screening in PCD.
Whole exome sequencing and array-based molecular karyotyping as aids to prenatal diagnosis in fetuses with suspected Simpson-Golabi-Behmel syndrome.
Simpson-Golabi-Behmel (SGBS) syndrome type 1 and type 2 represent rare X-linked prenatal overgrowth disorders. The aim of our study is to describe the prenatal sonographic features as well as the genetic work-up. Retrospective analysis of four cases with a pre- or postnatal diagnosis of SGBS in a single tertiary referral center within a period of 4 years. In the study period, four male fetuses with SGBS were detected. The final diagnosis was made prenatally in three cases. In all cases the second trimester anomaly scan revealed left sided congenital diaphragmatic hernia (CDH) with additional anomalies; three fetuses with SGBS type 1 showed fetal overgrowth. In two of these, whole exome sequencing showed a possible frameshift mutation and a point mutation in the gene GPC3, respectively. In the third case, multiplex ligation-dependent probe amplification (MLPA) revealed a hemizygous duplication of exon 3-7 in the gene GPC3. In the fourth case, SGBS type 2 was confirmed by array comparative genomic hybridization (CGH) of amniotic fluid cells showing a deletion of the gene OFD1. We could demonstrate, that in the presence of a CDH, syndromes of the fetus can be increasingly differentiated by detailed sonography followed by a selective and graded molecular diagnostic using microarray techniques and whole exome sequencing. © 2016 John Wiley & Sons, Ltd.
Publicações recentes
Expanding the Genotypic and Phenotypic Spectrum of OFD1-Related Conditions: Three More Cases.
The expanding phenotype of OFD1-related disorders: Hemizygous loss-of-function variants in three patients with primary ciliary dyskinesia.
Truncating mutations in exons 20 and 21 of OFD1 can cause primary ciliary dyskinesia without associated syndromic symptoms.
A recurrent germline mutation in the PIGA gene causes Simpson-Golabi-Behmel syndrome type 2.
Novel mutations including deletions of the entire OFD1 gene in 30 families with type 1 orofaciodigital syndrome: a study of the extensive clinical variability.
📚 EuropePMC131 artigos no totalmostrando 6
Expanding the Genotypic and Phenotypic Spectrum of OFD1-Related Conditions: Three More Cases.
GenesOFD Type I syndrome: lessons learned from a rare ciliopathy.
Biochemical Society transactionsThe expanding phenotype of OFD1-related disorders: Hemizygous loss-of-function variants in three patients with primary ciliary dyskinesia.
Molecular genetics & genomic medicineTruncating mutations in exons 20 and 21 of OFD1 can cause primary ciliary dyskinesia without associated syndromic symptoms.
Journal of medical geneticsWhole exome sequencing and array-based molecular karyotyping as aids to prenatal diagnosis in fetuses with suspected Simpson-Golabi-Behmel syndrome.
Prenatal diagnosisA recurrent germline mutation in the PIGA gene causes Simpson-Golabi-Behmel syndrome type 2.
American journal of medical genetics. Part AAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de Simpson-Golabi-Behmel tipo 2.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de Simpson-Golabi-Behmel tipo 2
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Expanding the Genotypic and Phenotypic Spectrum of OFD1-Related Conditions: Three More Cases.
- OFD Type I syndrome: lessons learned from a rare ciliopathy.
- The expanding phenotype of OFD1-related disorders: Hemizygous loss-of-function variants in three patients with primary ciliary dyskinesia.
- Truncating mutations in exons 20 and 21 of OFD1 can cause primary ciliary dyskinesia without associated syndromic symptoms.
- Whole exome sequencing and array-based molecular karyotyping as aids to prenatal diagnosis in fetuses with suspected Simpson-Golabi-Behmel syndrome.
- A recurrent germline mutation in the PIGA gene causes Simpson-Golabi-Behmel syndrome type 2.
- Novel mutations including deletions of the entire OFD1 gene in 30 families with type 1 orofaciodigital syndrome: a study of the extensive clinical variability.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:79022(Orphanet)
- OMIM OMIM:300209(OMIM)
- MONDO:0010265(MONDO)
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55999497(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome de Simpson-Golabi-Behmel tipo 2
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata