A síndrome de Mayer-Rokitansky-Küster-Hauser (MRKH) tipo 1, uma forma de síndrome MRKH, é uma forma isolada de aplasia congênita do útero e 2/3 da vagina que ocorre em mulheres fenotipicamente normais.
Introdução
O que você precisa saber de cara
A síndrome de Mayer-Rokitansky-Küster-Hauser (MRKH) tipo 1, uma forma de síndrome MRKH, é uma forma isolada de aplasia congênita do útero e 2/3 da vagina que ocorre em mulheres fenotipicamente normais.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 11 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 18 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Mayer-Rokitansky-Küster-Hauser clássico
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Mostrando amostra de 57 publicações de um total de 528
Variant phenotypic expression of mosaic Turner syndrome with type II MRKH (MURCS association).
We present a rare case of an unmarried woman in her mid-twenties with primary amenorrhoea who was found to have the unusual coexistence of mosaic Turner syndrome (45, XO/46, XX) and Mayer-Rokitansky-Küster-Hauser syndrome with MURCS association (Müllerian agenesis, Renal anomalies, Cervicothoracic Somite dysplasia). The patient exhibited normal secondary sexual characteristics and was phenotypically female. The patient underwent successful laparoscopic Davydov vaginoplasty in anticipation of marriage. This case highlights the diagnostic complexity and management challenges associated with dual congenital anomalies that affect both gonadal and Müllerian development.
A rare case of Mayer-Rokitansky-Küster-Hauser syndrome presenting with primary amenorrhea and chronic headaches: a case report.
Primary amenorrhea is defined as the absence of menarche by age 13 without secondary sexual characteristics or by age 15 with them. Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome is the second most common cause of primary amenorrhea after ovarian failure. This case highlights a 15-year-old female with MRKH syndrome type I presenting with chronic headaches. A 15-year-old female presented with primary amenorrhea and chronic frontal headaches. She reported non-foul-smelling white per vaginal discharge but denied visual disturbances, limb weakness, or systemic symptoms. Examination revealed normal secondary sexual characteristics, a blind vaginal pouch (0.5 cm), and a low body mass index of 17. Pelvic ultrasonography and MRI confirmed uterine agenesis with normal ovaries and no renal anomalies, consistent with MRKH syndrome. Brain MRI and hormonal profiles were normal, suggesting her headaches were likely nutritional and dehydration related, which improved with dietary optimization. A multidisciplinary team performed McIndoe vaginoplasty using a split-thickness skin graft, successfully creating a 9-cm neovagina. On follow-up, the patient demonstrated good neovaginal patency, maintained regular self-dilatation, and reported improved psychological well-being. MRKH syndrome is a congenital disorder characterized by the absence or underdevelopment of the uterus and upper vagina despite normal external genitalia and a 46,XX karyotype. It affects approximately 1 in 5000 females. Diagnosis is confirmed through imaging, with MRI being the most reliable modality. Although headaches are not commonly associated with MRKH, in this patient, they were likely due to undernutrition, dehydration, and psychological stress. Management of MRKH includes both nonsurgical (vaginal dilation) and surgical approaches. The McIndoe procedure, involving a skin graft to create a neovagina, has shown high success rates in improving sexual function and psychological well-being. Postoperative outcomes highlight the importance of a multidisciplinary approach. This case highlights a rare presentation of MRKH syndrome with primary amenorrhea and headache in a young adolescent. Although headaches may mislead diagnosis, imaging is vital for confirmation and exclusion of other causes. The patient improved following McIndoe vaginoplasty, emphasizing timely diagnosis, multidisciplinary care, and psychological support for optimal outcomes.
Prevalence, clinical profile, and associated anomalies with women with Mayer-Rokitansky-Küster-Hauser syndrome in a tertiary care center: A cross-sectional study.
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome is a rare congenital anomaly of the Müllerian ducts and represents the second most common cause of primary amenorrhea, accounting for 10%-15% of cases. Despite its significance, limited data exist regarding its clinical profile and associated anomalies in the Indian population. This study aims to determine the prevalence of MRKH syndrome among women presenting with primary amenorrhea at a tertiary care center in South India and to describe their clinical profiles and associated anomalies using the Vagina Cervix Uterus Adnexa-associated Malformation (VCUAM) classification system. A retrospective study was conducted over 15 years (January 2008 to December 2022) including women diagnosed with MRKH syndrome based on inclusion criteria: primary amenorrhea, normal secondary sexual characteristics, 46-XX karyotype, and normal serum follicle-stimulating hormone levels. Data were extracted from medical records and analyzed using SPSS v25.0. Out of 340 women with primary amenorrhea, 181 (60%) were diagnosed with MRKH syndrome. The mean age at presentation was 21 years. The predominant complaint was non-attainment of menarche (66.8%), with 16.5% reporting cyclical abdominal pain. Type 1 MRKH was most common (78.9%), followed by Müllerian duct aplasia-renal agenesis-cervicothoracic somite dysplasia (MURCS) association (16.5%). Renal anomalies (15.5%) were the most frequent extragenital malformations. All women had vaginal and cervical agenesis (V5bC2b). Uterine anomalies included bilateral aplasia (89.1%), unilateral aplasia (0.6%), and hypoplasia (10.5%). MRKH syndrome is a significant cause of primary amenorrhea, with notable extragenital anomalies, especially renal. Systematic evaluation using the VCUAM classification enables comprehensive assessment, aiding in individualized and multidisciplinary care strategies.
Mayer-Rokitansky-Kuster-Hauser Syndrome: From Radiological Diagnosis to Further Challenges-Review and Update.
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome encompasses a range of Müllerian duct anomalies characterized by congenital absence of the uterus and the upper two-thirds of the vagina in young women who otherwise exhibit normal endocrine function and a 46,XX karyotype. MRKH syndrome can occur in an isolated form (type I) or in association with other congenital anomalies (type II or MURCS association), which may include renal, vertebral, auditory, and cardiac defects. It represents one of the most frequent causes of primary amenorrhea, affecting approximately 1 in every 4000-5000 women. MRKH syndrome often remains undiagnosed until a patient presents with primary amenorrhea, despite normal development of secondary sexual characteristics. Both genetic and non-genetic factors have been proposed as contributing to abnormal embryonic development, although the exact etiopathogenesis remains unclear. Imaging plays a key role in the evaluation of genital tract anomalies, allowing non-invasive and comprehensive assessment. Alongside physical examination and pelvic ultrasound, pelvic MRI is essential for identifying the presence of rudimentary uterine tissue. MRKH syndrome can have profound and lasting psychological impacts, making it essential for patients and their families to receive counseling both before and throughout treatment. A range of therapeutic options-both surgical and non-surgical-have been proposed for managing MRKH syndrome. Vaginal dilation remains the first-line treatment, as it offers high success rates with minimal risk of complications. Vaginoplasty is considered a second-line option for patients who do not respond to dilation therapy. Additionally, uterine transplantation and gestational surrogacy provide opportunities for women with MRKH syndrome to achieve biological motherhood. This review provides an updated overview of Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome, encompassing its etiological, clinical, diagnostic, psychological, therapeutic, and reproductive aspects. We also present a case involving a 19-year-old woman with MRKH syndrome who presented with primary amenorrhea, highlighting the crucial role and advantages of MRI in diagnosis, differential assessment, and treatment planning.
Combined Vaginal and Laparoscopic Approach for the Creation of Neovagina in a Patient Affected by Mayer-Rokitansky-Küster-Hauser Syndrome: An Innovative Surgical Treatment.
To describe the step-by-step simultaneous vaginal and laparoscopic approach for the creation and reconstruction of a neovagina using a skin graft in a woman affected by Mayer-Rokitansky-Küster-Hauser syndrome [1]. To date, no data are available in the literature comparing our technique with other validated approaches such as McIndoe and Davydov, as our process is a newly developed one. Tertiary-referral center. An eighteen-year-old woman affected by type 1 Mayer-Rokitansky-Küster-Hauser syndrome previously treated with vaginal dilators without success attracted our attention because of her desire to treat her vaginal agenesis. The patient report primary amenorrhea and appeared with normal secondary sexual characteristics. Blind vaginal pouch was confirmed via imaging [2]. After having an accurate counseling with the patient, using drawings on the thigh, informing her about the postoperative course, and showing aesthetic outcomes, an innovative surgery, resulting from a combination of vaginal and laparoscopic approaches, was proposed. The surgical procedure consisted of 2 phases. The vaginal step involved the creation of the lower part of the neovagina using the Fortunoff technique, whereas the laparoscopic step enabled the creation of the upper part under direct visual control. The neovagina was reconstructed using a skin graft harvested from the patient's thigh. The combination of techniques enabled the development of a functional vaginal canal with excellent anatomical and clinical outcomes. At three-month follow-up, the neovagina appeared fully epithelialized, with a total length of 8 cm and satisfactory anatomical and functional results [3-5]. In conclusion, this is the first reported case of a technique combining a vaginal approach for creating the lower canal of the neovagina with a laparoscopic approach for the upper part. We are committed to increasing our case series so that our surgical approach may be considered a viable option to offer surgeons in the future. VIDEO ABSTRACT.
Publicações recentes
A Cognitive Behavioural Therapy for Mayer-Rokitansky-Küster-Hauser Syndrome Adapted for Other Disorders of Sex Development: A Qualitative Study.
Mayer-Rokitansky-Küster-Hauser Syndrome Associated With Diabetes Mellitus and Renal Anomalies in an Adolescent Girl: A Rare Case Report.
Regarding "Combined Vaginal and Laparoscopic Approach for the Creation of Neovagina in a Patient Affected by Mayer-Rokitansky-Küster-Hauser Syndrome: An Innovative Surgical Treatment".
Functional and anatomical results following combined vaginal and laparoscopic approach for the creation of neovagina in a patient affected by Mayer-Rokitansky-Küster-Hauser syndrome.
Anesthetic challenges of the first successful living-donor uterus transplantation in Latin America: a case report.
📚 EuropePMC574 artigos no totalmostrando 57
A rare case of Mayer-Rokitansky-Küster-Hauser syndrome presenting with primary amenorrhea and chronic headaches: a case report.
International journal of surgery case reportsPrevalence, clinical profile, and associated anomalies with women with Mayer-Rokitansky-Küster-Hauser syndrome in a tertiary care center: A cross-sectional study.
International journal of gynaecology and obstetrics: the official organ of the International Federation of Gynaecology and ObstetricsMayer-Rokitansky-Kuster-Hauser Syndrome: From Radiological Diagnosis to Further Challenges-Review and Update.
Diagnostics (Basel, Switzerland)Variant phenotypic expression of mosaic Turner syndrome with type II MRKH (MURCS association).
BMJ case reportsA Rare Case of Müllerian Agenesis With a Giant Tumor Arising From Uterine Remnants.
Case reports in obstetrics and gynecologyCondylomata acuminata in the neovagina and vulva with HPV6/18 infection in a patient with Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome after sheares' vaginoplasty: a case report and systematic review.
BMC women's healthSpinal Anomalies in MURCS Association: A Rare Case Report and Systematic Review of the Literature.
Congenital anomaliesCombined Vaginal and Laparoscopic Approach for the Creation of Neovagina in a Patient Affected by Mayer-Rokitansky-Küster-Hauser Syndrome: An Innovative Surgical Treatment.
Journal of minimally invasive gynecologyMcIndoe Vaginoplasty in MRKHS: Case Report and Literature Review.
Clinical case reportsOptimizing care for MRKH patients: From malformation screening to uterus transplantation eligibility.
Acta obstetricia et gynecologica ScandinavicaA case study of transneovaginal oocyte retrieval after novel Lee's neovaginoplasty in Mayer-Rokitansky-Küster-Hauser syndrome.
Taiwanese journal of obstetrics & gynecologyA Case of Chromosome 17q12 Deletion Syndrome with Type 2 Mayer-Rokitansky-Küster-Hauser Syndrome and Maturity-Onset Diabetes of the Young Type 5.
Children (Basel, Switzerland)Bilateral indirect ovarian inguinal hernia in a young female with type 1 Mayer-Rokitansky-Küster-Hauser syndrome: An extremely rare clinical context.
Clinical case reportsClinical features and management of women with Mayer-Rokitansky-Küster-Hauser syndrome in a Thai population.
Obstetrics & gynecology scienceA case report of laparoscopic surgery for Mayer-Rokitansky-Küster-Hauser syndrome with preservation of functional primordial uterus.
BMC women's healthSuccessful Use of Acellular Small Intestinal Submucosa Graft in Vaginal Reconstruction.
Journal of pediatric surgeryRecurrent human 16p11.2 microdeletions in type I Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome patients in Chinese Han population.
Molecular genetics & genomic medicineMüllerian Agenesis in a patient with Rubinstein-Taybi Syndrome: A Case Series and Review of the Overlapping Developmental Biologic Pathways.
Journal of pediatric and adolescent gynecologyHuge Leiomyomas Arising from Bilateral Uterine Remnants in a Mayer-Rokitansky-Küster-Hauser Syndrome Patient with Coexisting Myotonic Dystrophy Type 1: A Case Report and Literature Review.
Case reports in obstetrics and gynecologyRare variant enrichment analysis supports GREB1L as a contributory driver gene in the etiology of Mayer-Rokitansky-Küster-Hauser syndrome.
HGG advancesAn unusual association of type II Mayer-Rokitansky-Kuster-Hauser syndrome, turner mosaic syndrome and tubo-ovarian inguinal hernia- case report and review of literature.
Journal of ovarian researchRetrospective comparative cohort study of neovagina creation by modified Vecchietti-laparoendoscopic single-site surgery for Mayer-Rokitansky-Küster-Hauser syndrome.
Annals of translational medicineFunctional genomics analysis identifies loss of HNF1B function as a cause of Mayer-Rokitansky-Küster-Hauser syndrome.
Human molecular geneticsMayer-Rokitansky-Kuster-Hauser Syndrome: A rare case report from Nepal.
Annals of medicine and surgery (2012)Clinical features of Mayer-Rokitansky-Küster-Haüser syndrome diagnosed at under 16 years old: results from a questionnaire survey conducted on all institutions of pediatric surgery and pediatric urology in Japan.
Pediatric surgery internationalRenal abnormalities associated with Mayer-Rokitansky-Küster-Hauser syndrome.
Folia medicaTissue Engineering Neovagina for Vaginoplasty in Mayer-Rokitansky-Küster-Hauser Syndrome and Gender Dysphoria Patients: A Systematic Review.
Tissue engineering. Part B, ReviewsVariants in genes related to development of the urinary system are associated with Mayer-Rokitansky-Küster-Hauser syndrome.
Human genomicsUtero-Ovarian Inguinal Hernia in a Young Female with Mayer-Rokitansky-Küster-Hauser Syndrome Type 2.
Saudi journal of medicine & medical sciencesPrimary Amenorrhea Due to Anatomical Abnormalities of the Reproductive Tract: Molecular Insight.
International journal of molecular sciencesOvarian inguinal hernia - a possibility in MURCS syndrome.
Journal of ovarian researchImplications of Ehlers-Danlos Syndrome in a Patient With Mayer-Rokitansky-Küster-Hauser Syndrome.
Journal of pediatric and adolescent gynecologyClinical characteristics of 1,055 Chinese patients with Mayer-Rokitansky-Küster-Hauser syndrome: a nationwide multicentric study.
Fertility and sterilityGREB1L as a candidate gene of Mayer-Rokitansky-Küster-Hauser Syndrome.
European journal of medical geneticsMayer-Rokitansky-Küster-Hauser (MRKH) syndrome: a comprehensive update.
Orphanet journal of rare diseasesMayer-Rokitansky-Küster-Hauser syndrome as an interdisciplinary problem.
Advances in clinical and experimental medicine : official organ Wroclaw Medical UniversityNeovagina Creation: A Novel Improved Laparoscopic Vecchietti Procedure in Patients with Mayer-Rokitansky-Küster-Hauster Syndrome.
Journal of minimally invasive gynecologyLive birth after robotic-assisted live donor uterus transplantation.
Acta obstetricia et gynecologica ScandinavicaDetection of de novo genetic variants in Mayer-Rokitansky-Küster-Hauser syndrome by whole genome sequencing.
European journal of obstetrics & gynecology and reproductive biology: XEvaluation of the abdominopelvic region using MRI in patients with primary amenorrhea.
Journal of pediatric endocrinology & metabolism : JPEMIdentification of Candidate Genes for Mayer-Rokitansky-Küster-Hauser Syndrome Using Genomic Approaches.
Sexual development : genetics, molecular biology, evolution, endocrinology, embryology, and pathology of sex determination and differentiationSpectrum of Type I and Type II Syndromes and Associated Malformations in Chinese Patients with Mayer-Rokitansky-Küster-Hauser Syndrome: A Retrospective Analysis of 274 Cases.
Journal of pediatric and adolescent gynecologyVaginal prevalence of human papillomavirus infections in women with uterovaginal aplasia before and after laparoscopically assisted creation of a neovagina: a prospective epidemiological observational study.
BJOG : an international journal of obstetrics and gynaecologyClinical and genetic aspects of Mayer-Rokitansky-Küster-Hauser syndrome.
Medizinische Genetik : Mitteilungsblatt des Berufsverbandes Medizinische Genetik e.VHyperandrogenemia and ovarian reserve in patients with Mayer-Rokitansky-Küster-Hauser syndrome type 1 and 2: potential influences on ovarian stimulation.
Archives of gynecology and obstetricsObstructive Müllerian Anomalies in Menstruating Adolescent Girls: A Report of 22 Cases.
Journal of pediatric and adolescent gynecologyGenetic analysis of Mayer-Rokitansky-Kuster-Hauser syndrome in a large cohort of families.
Fertility and sterilityLiving-Donor Kidney Transplant in a Patient With Type B Mayer-Rokitansky-Küster-Hauser Syndrome, Reconstructed Vagina, and Abnormal Pelvic Vessels: A Case Report.
Experimental and clinical transplantation : official journal of the Middle East Society for Organ TransplantationCongenital malformations and other comorbidities in 125 women with Mayer-Rokitansky-Küster-Hauser syndrome.
European journal of obstetrics, gynecology, and reproductive biologyGenetics of Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome.
Clinical geneticsMutations in WNT9B are associated with Mayer-Rokitansky-Küster-Hauser syndrome.
Clinical geneticsAetiological bases of 46,XY disorders of sex development in the Hong Kong Chinese population.
Hong Kong medical journal = Xianggang yi xue za zhiTypical and Atypical Associated Findings in a Group of 346 Patients with Mayer-Rokitansky-Kuester-Hauser Syndrome.
Journal of pediatric and adolescent gynecologyAssociations of Polymorphisms in WNT9B and PBX1 with Mayer-Rokitansky-Küster-Hauser Syndrome in Chinese Han.
PloS one[Mayer-Rokitansky-Küster-Hauser syndrome: two cases report].
Ginecologia y obstetricia de MexicoComparison of markers of ovarian reserve between patients with complete müllerian agenesis and age-matched fertile and infertile controls.
Fertility and sterilityConcurrent exome-targeted next-generation sequencing and single nucleotide polymorphism array to identify the causative genetic aberrations of isolated Mayer-Rokitansky-Küster-Hauser syndrome.
Human reproduction (Oxford, England)Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Mayer-Rokitansky-Küster-Hauser clássico.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Mayer-Rokitansky-Küster-Hauser clássico
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Variant phenotypic expression of mosaic Turner syndrome with type II MRKH (MURCS association).
- A rare case of Mayer-Rokitansky-Küster-Hauser syndrome presenting with primary amenorrhea and chronic headaches: a case report.
- Prevalence, clinical profile, and associated anomalies with women with Mayer-Rokitansky-Küster-Hauser syndrome in a tertiary care center: A cross-sectional study.International journal of gynaecology and obstetrics: the official organ of the International Federation of Gynaecology and Obstetrics· 2026· PMID 41636313mais citado
- Mayer-Rokitansky-Kuster-Hauser Syndrome: From Radiological Diagnosis to Further Challenges-Review and Update.
- Combined Vaginal and Laparoscopic Approach for the Creation of Neovagina in a Patient Affected by Mayer-Rokitansky-Küster-Hauser Syndrome: An Innovative Surgical Treatment.
- A Cognitive Behavioural Therapy for Mayer-Rokitansky-Küster-Hauser Syndrome Adapted for Other Disorders of Sex Development: A Qualitative Study.
- Mayer-Rokitansky-Küster-Hauser Syndrome Associated With Diabetes Mellitus and Renal Anomalies in an Adolescent Girl: A Rare Case Report.
- Regarding "Combined Vaginal and Laparoscopic Approach for the Creation of Neovagina in a Patient Affected by Mayer-Rokitansky-Küster-Hauser Syndrome: An Innovative Surgical Treatment".
- Functional and anatomical results following combined vaginal and laparoscopic approach for the creation of neovagina in a patient affected by Mayer-Rokitansky-Küster-Hauser syndrome.
- Anesthetic challenges of the first successful living-donor uterus transplantation in Latin America: a case report.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:247775(Orphanet)
- OMIM OMIM:277000(OMIM)
- MONDO:0010173(MONDO)
- GARD:4737(GARD (NIH))
- Busca completa no PubMed(PubMed)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
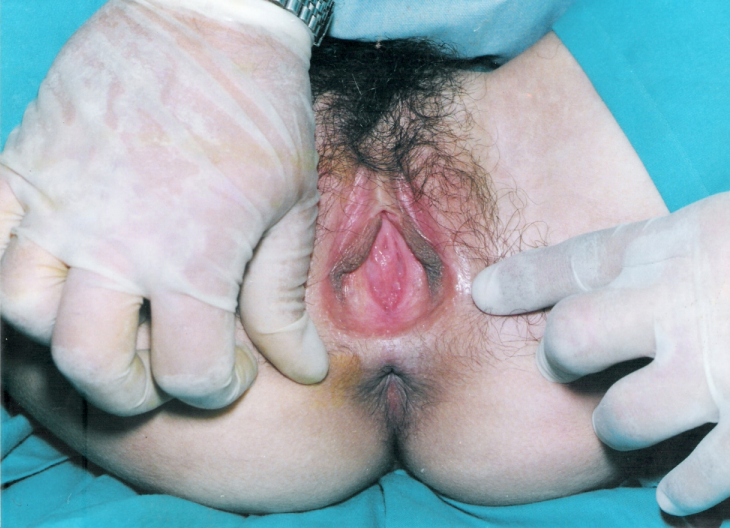
Síndrome Mayer-Rokitansky-Küster-Hauser clássico
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)