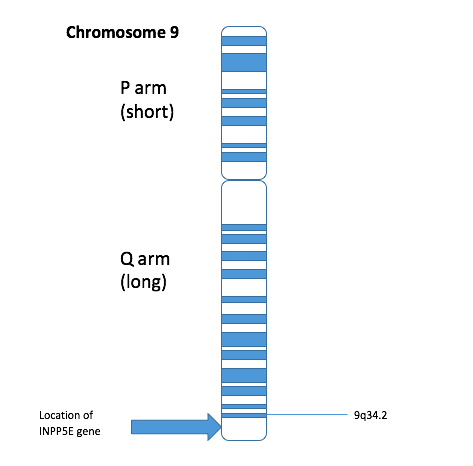
A síndrome MORM é caracterizada pela combinação de deficiência intelectual, acúmulo de gordura no tronco (obesidade truncal), um problema na retina que afeta a visão (distrofia retiniana) e pênis de tamanho reduzido (micropênis). Ela foi descrita em 14 pessoas de uma família onde os pais são parentes de sangue (consanguínea). A transmissão é autossômica recessiva, o que significa que a pessoa só desenvolve a síndrome se herdar um gene alterado tanto do pai quanto da mãe. O gene responsável pela síndrome foi localizado na região 9q34 de um cromossomo.
Introdução
O que você precisa saber de cara
A síndrome MORM é caracterizada pela combinação de deficiência intelectual, acúmulo de gordura no tronco (obesidade truncal), um problema na retina que afeta a visão (distrofia retiniana) e pênis de tamanho reduzido (micropênis). Ela foi descrita em 14 pessoas de uma família onde os pais são parentes de sangue (consanguínea). A transmissão é autossômica recessiva, o que significa que a pessoa só desenvolve a síndrome se herdar um gene alterado tanto do pai quanto da mãe. O gene responsável pela síndrome foi localizado na região 9q34 de um cromossomo.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 7 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 24 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisPhosphatidylinositol (PtdIns) phosphatase that specifically hydrolyzes the 5-phosphate of phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3), phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) and phosphatidylinositol 3,5-bisphosphate (PtdIns(3,5)P2) (By similarity) (PubMed:10764818). Specific for lipid substrates, inactive towards water soluble inositol phosphates (PubMed:10764818). Plays an essential role in the primary cilium by controlling ciliary growth and phosphoinositide 3-kin
Cytoplasm, cytoskeleton, cilium axonemeGolgi apparatus, Golgi stack membraneCell membraneCell projection, ruffleCytoplasmNucleus
Joubert syndrome 1
A disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy and renal disease.
Variantes genéticas (ClinVar)
216 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 123 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome MORM
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Novel Insights Into Monogenic Obesity Syndrome Due to INPP5E Gene Variant: A Case Report of a Female Patient.
A Caucasian girl with consanguineous parents presented with early severe obesity and retinal dystrophy. A novel, homozygous gene truncating variant (c.1897C>T) in the INPP5E gene confirmed the diagnosis of MORMS (OMIM #610156). A novel clinical finding in the presented syndrome is progressive cone-rod type retinal dystrophy diagnosed at the age of four months that progressed in the 1st decade of life. Severe obesity, insulin resistance with hyperinsulinism, and impaired glucose tolerance developed alongside other components of the metabolic syndrome - dyslipidemia, arterial hypertension, and obstructive hypopnea in sleep. At the age of 14 years, primary amenorrhea persists. The patient is managed by regular nutritional advice, metformin, antihypertensive medication, and non-invasive respiratory support during sleep. Differential diagnosis of this rare entity is discussed in extend.
BBS5 and INPP5E mutations associated with ciliopathy disorders in families from Pakistan.
Ciliopathies are a clinically and genetically heterogeneous group of disorders often exhibiting phenotypic overlap and caused by abnormalities in the structure or function of cellular cilia. As such, a precise molecular diagnosis is important for guiding clinical management and genetic counseling. In the present study, two Pakistani families comprising individuals with overlapping clinical features suggestive of a ciliopathy syndrome, including intellectual disability, obesity, congenital retinal dystrophy, and hypogonadism (in males), were investigated clinically and genetically. Whole-exome sequencing identified the likely causes of disease as a novel homozygous frameshift mutation (NM_152384.2: c.196delA; p.(Arg66Glufs*12); family 1) in BBS5, and a nonsense mutation (NM_019892.5:c.1879C>T; p.Gln627*; family 2) in INPP5E, previously reported in an extended Pakistani family with MORM syndrome. Our findings expand the molecular spectrum associated with BBS5 mutations in Pakistan and provide further supportive evidence that the INPP5E mutation is a common cause of ciliopathy in Northern Pakistan, likely representing a regional founder mutation. This study also highlights the value of genomic studies in Pakistan for families affected by rare heterogeneous developmental disorders and where clinical phenotyping may be limited by geographical and financial constraints. The identification of the spectrum and frequency of disease-causing variants within this setting enables the development of population-specific genetic testing strategies targeting variants common to the local population and improving health care outcomes.
Role of reverse phenotyping in interpretation of next generation sequencing data and a review of INPP5E related disorders.
Next Generation Sequencing (NGS) is a useful tool in diagnosis of rare disorders but the interpretation of data can be challenging in clinical settings. We present results of extended studies on a family of multiple members with global developmental delay and learning disability, where another research group postulated the underlying cause to be a homozygous RABL6 missense variant. Using data from the Exome Variant Server, we show that missense RABL6 variants are unlikely to cause early onset rare developmental disorder. Protein structural analysis, cellular functional studies and reverse phenotyping proved that the condition in this family is due to a homozygous INPP5E mutation. An in-depth review of mutational and phenotypic spectrum associated with INPP5E demonstrated that mutations in this gene lead to a range of cilliopathy-phenotypes. We use this study as an example to demonstrate the importance of careful clinical evaluation of multiple family members, reverse phenotyping, considering the unknown phenotypic variability of rare diseases, utilizing publically available genomic databases and conducting appropriate bioinformatics and functional studies while interpreting results from NGS in uncertain cases. We emphasize that interpretation of NGS data is an iterative process and its dynamic nature should be explained to patients and families. Our study shows that developmental delay, intellectual disability, hypotonia and ocular motor apraxia are common in INPP5E-related disorders and considerable intra-familial phenotypic variability is possible. We have compiled the INPP5E mutational spectrum and provided novel insights into their molecular mechanisms.
Modulation of Ciliary Phosphoinositide Content Regulates Trafficking and Sonic Hedgehog Signaling Output.
Ciliary transport is required for ciliogenesis, signal transduction, and trafficking of receptors to the primary cilium. Mutations in inositol polyphosphate 5-phosphatase E (INPP5E) have been associated with ciliary dysfunction; however, its role in regulating ciliary phosphoinositides is unknown. Here we report that in neural stem cells, phosphatidylinositol 4-phosphate (PI4P) is found in high levels in cilia whereas phosphatidylinositol (4,5)-bisphosphate (PI(4,5)P2) is not detectable. Upon INPP5E inactivation, PI(4,5)P2 accumulates at the ciliary tip whereas PI4P is depleted. This is accompanied by recruitment of the PI(4,5)P2-interacting protein TULP3 to the ciliary membrane, along with Gpr161. This results in an increased production of cAMP and a repression of the Shh transcription gene Gli1. Our results reveal the link between ciliary regulation of phosphoinositides by INPP5E and Shh regulation via ciliary trafficking of TULP3/Gpr161 and also provide mechanistic insight into ciliary alterations found in Joubert and MORM syndromes resulting from INPP5E mutations.
Publicações recentes
BBS5 and INPP5E mutations associated with ciliopathy disorders in families from Pakistan.
Role of reverse phenotyping in interpretation of next generation sequencing data and a review of INPP5E related disorders.
INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse.
MORM syndrome (mental retardation, truncal obesity, retinal dystrophy and micropenis), a new autosomal recessive disorder, links to 9q34.
📚 EuropePMC1 artigos no totalmostrando 4
Novel Insights Into Monogenic Obesity Syndrome Due to INPP5E Gene Variant: A Case Report of a Female Patient.
Frontiers in endocrinologyBBS5 and INPP5E mutations associated with ciliopathy disorders in families from Pakistan.
Annals of human geneticsRole of reverse phenotyping in interpretation of next generation sequencing data and a review of INPP5E related disorders.
European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology SocietyModulation of Ciliary Phosphoinositide Content Regulates Trafficking and Sonic Hedgehog Signaling Output.
Developmental cellAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome MORM.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome MORM
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Novel Insights Into Monogenic Obesity Syndrome Due to INPP5E Gene Variant: A Case Report of a Female Patient.
- BBS5 and INPP5E mutations associated with ciliopathy disorders in families from Pakistan.
- Role of reverse phenotyping in interpretation of next generation sequencing data and a review of INPP5E related disorders.European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology Society· 2016· PMID 26748598mais citado
- Modulation of Ciliary Phosphoinositide Content Regulates Trafficking and Sonic Hedgehog Signaling Output.
- INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse.
- MORM syndrome (mental retardation, truncal obesity, retinal dystrophy and micropenis), a new autosomal recessive disorder, links to 9q34.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:75858(Orphanet)
- OMIM OMIM:610156(OMIM)
- MONDO:0012423(MONDO)
- GARD:10121(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q6717114(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome MORM
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata