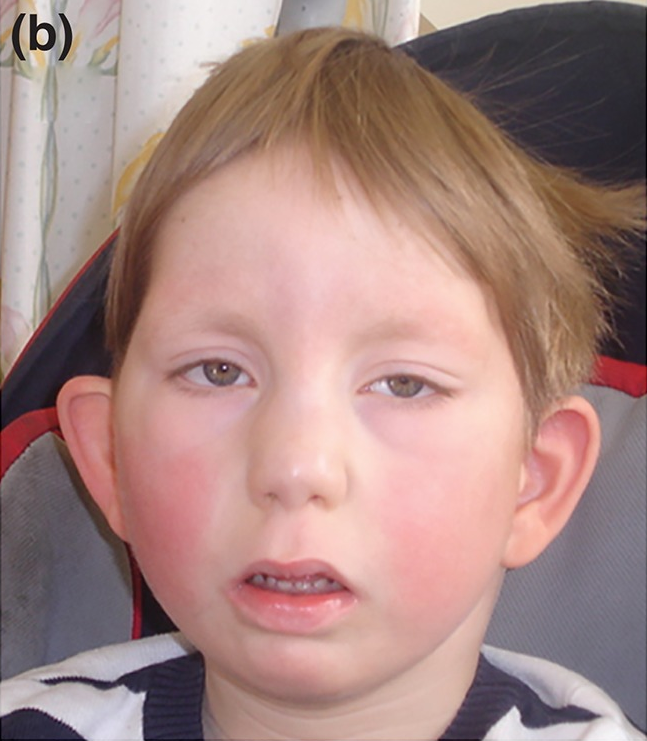
Síndrome rara associada a mutações no gene HNRNPK, caracterizada por refluxo vesicoureteral, defeitos cardíacos (comunicação interventricular), anomalias craniofaciais (ponte nasal ampla, craniossinostose lambdóide) e ortopédicas (pé plano, pé torto). Pode requerer alimentação por sonda e apresentar mordida aberta e eversão palpebral.
Introdução
O que você precisa saber de cara
Visão geral
A Síndrome Okamuto (também conhecida como Síndrome Okamoto) é uma condição genética rara que afeta múltiplos sistemas do corpo. As pessoas com essa síndrome podem apresentar uma combinação de características que incluem alterações cardíacas, renais, craniofaciais, neurológicas e musculoesqueléticas. A condição é causada por alterações (variantes) no gene HNRNPK.[1][2][4]
Sinais e sintomas
Os sinais e sintomas da Síndrome Okamuto podem variar de pessoa para pessoa, mas frequentemente incluem: defeitos cardíacos como o defeito do septo ventricular; problemas renais como refluxo vesicoureteral; características faciais como ponte nasal ampla, eversão do terço lateral das pálpebras inferiores, fossa pré-auricular, orelhas proeminentes e macroglossia (língua aumentada); alterações neurológicas como deficiência intelectual grave, heterotopia de substância cinzenta, fraqueza muscular, reflexos tendíneos reduzidos ou arreflexia; dificuldades alimentares na infância que podem exigir alimentação por sonda nasogástrica; problemas gastrointestinais como dismotilidade gastrointestinal e gastroparesia; além de miopia, deficiência auditiva condutiva, craniossinostose lambdóide, pé plano, pé torto equinovaro, mordida aberta, fadiga fácil, atraso de crescimento e aumento da translucência nucal.[1][4]
Causas genéticas
A Síndrome Okamuto é causada por alterações (variantes patogênicas) no gene HNRNPK (sigla em inglês para Heterogeneous nuclear ribonucleoprotein K). Esse gene fornece instruções para a produção de uma proteína que participa de diversos processos celulares importantes, incluindo a regulação da expressão de outros genes. As variantes nesse gene alteram a função da proteína, levando aos sinais e sintomas observados na síndrome.[1][2][5]
Diagnóstico
O diagnóstico da Síndrome Okamuto é baseado na avaliação clínica dos sinais e sintomas característicos e confirmado por meio de teste genético molecular que identifique uma variante patogênica no gene HNRNPK. Atualmente, há 148 variantes registradas no ClinVar (um banco de dados público de variantes genéticas e sua relação com doenças) associadas a essa condição.[1][2][5]
Tratamento e manejo
Não existe cura para a Síndrome Okamuto, e o tratamento é focado no manejo dos sintomas e na melhora da qualidade de vida. O cuidado geralmente envolve uma equipe multidisciplinar, incluindo cardiologista, nefrologista, neurologista, geneticista, fonoaudiólogo, fisioterapeuta e outros especialistas, conforme a necessidade de cada pessoa. Intervenções podem incluir cirurgia para correção de defeitos cardíacos ou renais, terapia para dificuldades alimentares, suporte educacional para deficiência intelectual, e fisioterapia para fraqueza muscular e alterações ortopédicas. Não há medicamentos específicos aprovados para a síndrome.[1][2]
Prognóstico e qualidade de vida
O prognóstico para pessoas com Síndrome Okamuto varia amplamente, dependendo da gravidade dos sintomas e do envolvimento de órgãos vitais, como coração e rins. O acompanhamento médico regular e o manejo adequado das complicações podem melhorar significativamente a qualidade de vida e a sobrevida. O suporte familiar e o acesso a terapias multidisciplinares são fundamentais.[1][2]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Síndrome rara associada a mutações no gene HNRNPK, caracterizada por refluxo vesicoureteral, defeitos cardíacos (comunicação interventricular), anomalias craniofaciais (ponte nasal ampla, craniossinostose lambdóide) e ortopédicas (pé plano, pé torto). Pode requerer alimentação por sonda e apresentar mordida aberta e eversão palpebral.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Síndrome Okamuto (também conhecida como Síndrome Okamoto) é uma condição genética rara que afeta múltiplos sistemas do corpo. As pessoas com essa síndrome podem apresentar uma combinação de características que incluem alterações cardíacas, renais, craniofaciais, neurológicas e musculoesqueléticas. A condição é causada por alterações (variantes) no gene HNRNPK.[1][2][4]
Sinais e sintomas
Os sinais e sintomas da Síndrome Okamuto podem variar de pessoa para pessoa, mas frequentemente incluem: defeitos cardíacos como o defeito do septo ventricular; problemas renais como refluxo vesicoureteral; características faciais como ponte nasal ampla, eversão do terço lateral das pálpebras inferiores, fossa pré-auricular, orelhas proeminentes e macroglossia (língua aumentada); alterações neurológicas como deficiência intelectual grave, heterotopia de substância cinzenta, fraqueza muscular, reflexos tendíneos reduzidos ou arreflexia; dificuldades alimentares na infância que podem exigir alimentação por sonda nasogástrica; problemas gastrointestinais como dismotilidade gastrointestinal e gastroparesia; além de miopia, deficiência auditiva condutiva, craniossinostose lambdóide, pé plano, pé torto equinovaro, mordida aberta, fadiga fácil, atraso de crescimento e aumento da translucência nucal.[1][4]
Causas genéticas
A Síndrome Okamuto é causada por alterações (variantes patogênicas) no gene HNRNPK (sigla em inglês para Heterogeneous nuclear ribonucleoprotein K). Esse gene fornece instruções para a produção de uma proteína que participa de diversos processos celulares importantes, incluindo a regulação da expressão de outros genes. As variantes nesse gene alteram a função da proteína, levando aos sinais e sintomas observados na síndrome.[1][2][5]
Diagnóstico
O diagnóstico da Síndrome Okamuto é baseado na avaliação clínica dos sinais e sintomas característicos e confirmado por meio de teste genético molecular que identifique uma variante patogênica no gene HNRNPK. Atualmente, há 148 variantes registradas no ClinVar (um banco de dados público de variantes genéticas e sua relação com doenças) associadas a essa condição.[1][2][5]
Tratamento e manejo
Não existe cura para a Síndrome Okamuto, e o tratamento é focado no manejo dos sintomas e na melhora da qualidade de vida. O cuidado geralmente envolve uma equipe multidisciplinar, incluindo cardiologista, nefrologista, neurologista, geneticista, fonoaudiólogo, fisioterapeuta e outros especialistas, conforme a necessidade de cada pessoa. Intervenções podem incluir cirurgia para correção de defeitos cardíacos ou renais, terapia para dificuldades alimentares, suporte educacional para deficiência intelectual, e fisioterapia para fraqueza muscular e alterações ortopédicas. Não há medicamentos específicos aprovados para a síndrome.[1][2]
Prognóstico e qualidade de vida
O prognóstico para pessoas com Síndrome Okamuto varia amplamente, dependendo da gravidade dos sintomas e do envolvimento de órgãos vitais, como coração e rins. O acompanhamento médico regular e o manejo adequado das complicações podem melhorar significativamente a qualidade de vida e a sobrevida. O suporte familiar e o acesso a terapias multidisciplinares são fundamentais.[1][2]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 52 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 132 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição.
One of the major pre-mRNA-binding proteins. Binds tenaciously to poly(C) sequences. Likely to play a role in the nuclear metabolism of hnRNAs, particularly for pre-mRNAs that contain cytidine-rich sequences. Can also bind poly(C) single-stranded DNA. Plays an important role in p53/TP53 response to DNA damage, acting at the level of both transcription activation and repression. When sumoylated, acts as a transcriptional coactivator of p53/TP53, playing a role in p21/CDKN1A and 14-3-3 sigma/SFN in
CytoplasmNucleus, nucleoplasmCell projection, podosome
Au-Kline syndrome
A disorder characterized by intellectual disability, facial dysmorphism, cardiac defects, and connective tissue and skeletal abnormalities. Dysmorphic features include long palpebral fissures, ptosis, a broad prominent nasal bridge, hypoplastic alae nasi, an open downturned mouth, ears with underdeveloped and thick helices, high palate, and a unique tongue with a prominent median crease. Hypotonia, hyporeflexia, and high pain tolerance are additional features.
Variantes genéticas (ClinVar)
148 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
6 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Okamuto
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
An HNRNPK-specific DNA methylation signature makes sense of missense variants and expands the phenotypic spectrum of Au-Kline syndrome.
Au-Kline syndrome (AKS) is a neurodevelopmental disorder associated with multiple malformations and a characteristic facial gestalt. The first individuals ascertained carried de novo loss-of-function (LoF) variants in HNRNPK. Here, we report 32 individuals with AKS (26 previously unpublished), including 13 with de novo missense variants. We propose new clinical diagnostic criteria for AKS that differentiate it from the clinically overlapping Kabuki syndrome and describe a significant phenotypic expansion to include individuals with missense variants who present with subtle facial features and few or no malformations. Many gene-specific DNA methylation (DNAm) signatures have been identified for neurodevelopmental syndromes. Because HNRNPK has roles in chromatin and epigenetic regulation, we hypothesized that pathogenic variants in HNRNPK may be associated with a specific DNAm signature. Here, we report a unique DNAm signature for AKS due to LoF HNRNPK variants, distinct from controls and Kabuki syndrome. This DNAm signature is also identified in some individuals with de novo HNRNPK missense variants, confirming their pathogenicity and the phenotypic expansion of AKS to include more subtle phenotypes. Furthermore, we report that some individuals with missense variants have an "intermediate" DNAm signature that parallels their milder clinical presentation, suggesting the presence of an epi-genotype phenotype correlation. In summary, the AKS DNAm signature may help elucidate the underlying pathophysiology of AKS. This DNAm signature also effectively supported clinical syndrome delineation and is a valuable aid for variant interpretation in individuals where a clinical diagnosis of AKS is unclear, particularly for mild presentations.
A second case of Okamoto syndrome caused by HNRNPK mutation.
Okamoto syndrome has features overlapping with Au-Kline syndrome and is caused by HNRNPK mutation.
Okamoto syndrome is characterized by severe intellectual disability, generalized hypotonia, stenosis of the ureteropelvic junction with hydronephrosis, cardiac anomalies, and characteristic facial gestalt. Several patients have been reported. The basic mechanism of Okamoto syndrome has not been clarified. Au-Kline syndrome is a new syndrome due to loss-of-function variants in the HNRNPK (heterogeneous nuclear ribonucleoprotein K) gene. A new patient with Okamoto syndrome visited our hospital. We noticed that the patient had features overlapping with Au-Kline syndrome. We studied the HNRNPK gene by Sanger sequencing, and identified a novel splicing variant. We suggest that Okamoto syndrome is identical to Au-Kline syndrome.
A new finding of a tethered cord in a patient with Okamoto syndrome.
Publicações recentes
An HNRNPK-specific DNA methylation signature makes sense of missense variants and expands the phenotypic spectrum of Au-Kline syndrome.
A second case of Okamoto syndrome caused by HNRNPK mutation.
Okamoto syndrome has features overlapping with Au-Kline syndrome and is caused by HNRNPK mutation.
A new finding of a tethered cord in a patient with Okamoto syndrome.
📚 EuropePMC7 artigos no totalmostrando 4
An HNRNPK-specific DNA methylation signature makes sense of missense variants and expands the phenotypic spectrum of Au-Kline syndrome.
American journal of human geneticsA second case of Okamoto syndrome caused by HNRNPK mutation.
American journal of medical genetics. Part AOkamoto syndrome has features overlapping with Au-Kline syndrome and is caused by HNRNPK mutation.
American journal of medical genetics. Part AA new finding of a tethered cord in a patient with Okamoto syndrome.
Clinical dysmorphologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Okamuto.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Okamuto
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- An HNRNPK-specific DNA methylation signature makes sense of missense variants and expands the phenotypic spectrum of Au-Kline syndrome.
- A second case of Okamoto syndrome caused by HNRNPK mutation.
- Okamoto syndrome has features overlapping with Au-Kline syndrome and is caused by HNRNPK mutation.
- A new finding of a tethered cord in a patient with Okamoto syndrome.
- Au-Kline Syndrome.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2729(Orphanet)
- OMIM OMIM:604916(OMIM)
- MONDO:0014700(MONDO)
- GARD:4064(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55783391(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome Okamuto
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata